A group of researchers in Spain methodically evaluated exome data from 10,000 tumors in an attempt to identify cancer predisposition genes as well as verify their statistical method. They created a statistical method called ALFRED, which stands for allelic loss featuring rare damaging. They used this method to evaluate Alfred Knudson’s classic “two hit” postulation which originated in 1971. The article was published July 4, 2018 in Nature Communications.
Knudson was a geneticist researching retinoblastoma and his interest focused on the loss of function mechanism based on the knowledge that most tumor suppressor genes are recessive in nature. This is what led him to propose the “two hit” hypothesis as a mechanism for the de novo development of cancer.
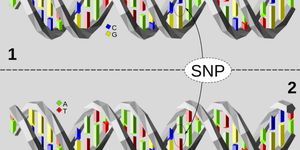
The ALFRED model utilizes a principal component analysis to cluster common variants in cancer patients. Then using two different test types, they quantify rare damaging germline variants with loss of heterozygosity events (termed allelic imbalance) compared to matched non-tumor samples from each patient which leads to an ALFRED P-value. ALFRED was applied to 10,043 tumor exomes which had sequencing data as part of The Cancer Genome Atlas project.
After evaluating the data against previously known cancer predisposition genes from recent literature reviews and the Cancer Gene Census, the group determined that these showed increased numbers of rare damaging germline variants with high frequency of allelic imbalance. They also evaluated somatic cancer genes for germline risk variants; their findings indicate that there are a few genes that are affected by somatic changes but may also further cancer development because of rare germline variations.
The research points to potential new germline cancer predisposition variants including NSD1, MYH1, NOP56, PRPF8, INO80, ANK2, and ATM.
More significant research is needed to explore the use of a statistical method like ALFRED to further classify these cancer predisposition genes, in addition to clinical tumor testing research to identify these genes in patients with specific cancers.
Sources: Nature Communications, Nature Education, The Cancer Genome Atlas,