The human genome project, which aimed to sequence the human genome in its entirety, was declared complete in 2003, and was celebrated as a major milestone for science. Genetic sequencing tools had gotten more sophisticated, efficient, and inexpensive, which helped make the achievement possible. But behind the scenes, there were some problems. The human genome contains many repetitive regions that don't code for genes and can be extremely challenging to sequence, so there were some gaps. Most of the work was also performed using a genomic sample from one individual, so it was an incomplete picture. The 1000 Genomes Project, completed in 2012, sequenced 1,092 genomes so that we would learn more about variation in the human genome. But that is still an underrepresentation and the data may not be included in reference sequences, potentially because of quality issues.
Last year, the National Institute of Health announced an initiative to address these problems. Called the “human pangenome reference,” the project aims to sequence the entire genomes of 350 individuals.
"One human genome cannot represent all of humanity. The human pangenome reference will be a key step forward for biomedical research and personalized medicine. Not only will we have 350 genomes representing human diversity, they will be vastly higher quality than previous genome sequences," said David Haussler, professor of biomolecular engineering at UC Santa Cruz and director of the UC Santa Cruz Genomics Institute.
Nobel Laureate Frederick Sanger and colleagues created Sanger sequencing for reading a genetic sequence in 1977. After a DNA sequence has been amplified, each base is labeled and then read from one end. In 1995, pairwise end sequencing was shown to be useful for sequencing whole genomes, and the technology was used to sequence the human genome. A library of fragments is prepared from a genome, and the fragments are read. Computational tools assemble the fragments into longer sequences. Nanopore sequencing, which pushes a molecule through a really tiny pore, detecting the base as it moves through, was created as one of the 'third-generation' sequencing technologies.
With an intensive effort, including 150,000 hours of commuting time, researchers at UC Santa Cruz researchers were able to use nanopore technology for long-read human genome sequencing. Around a year later, the cost was brought down significantly, and the results could were then obtained within about a week. "We sequenced eleven human genomes in nine days, which was unprecedented at the time," said UC Santa Cruz Research Scientist Miten Jain.
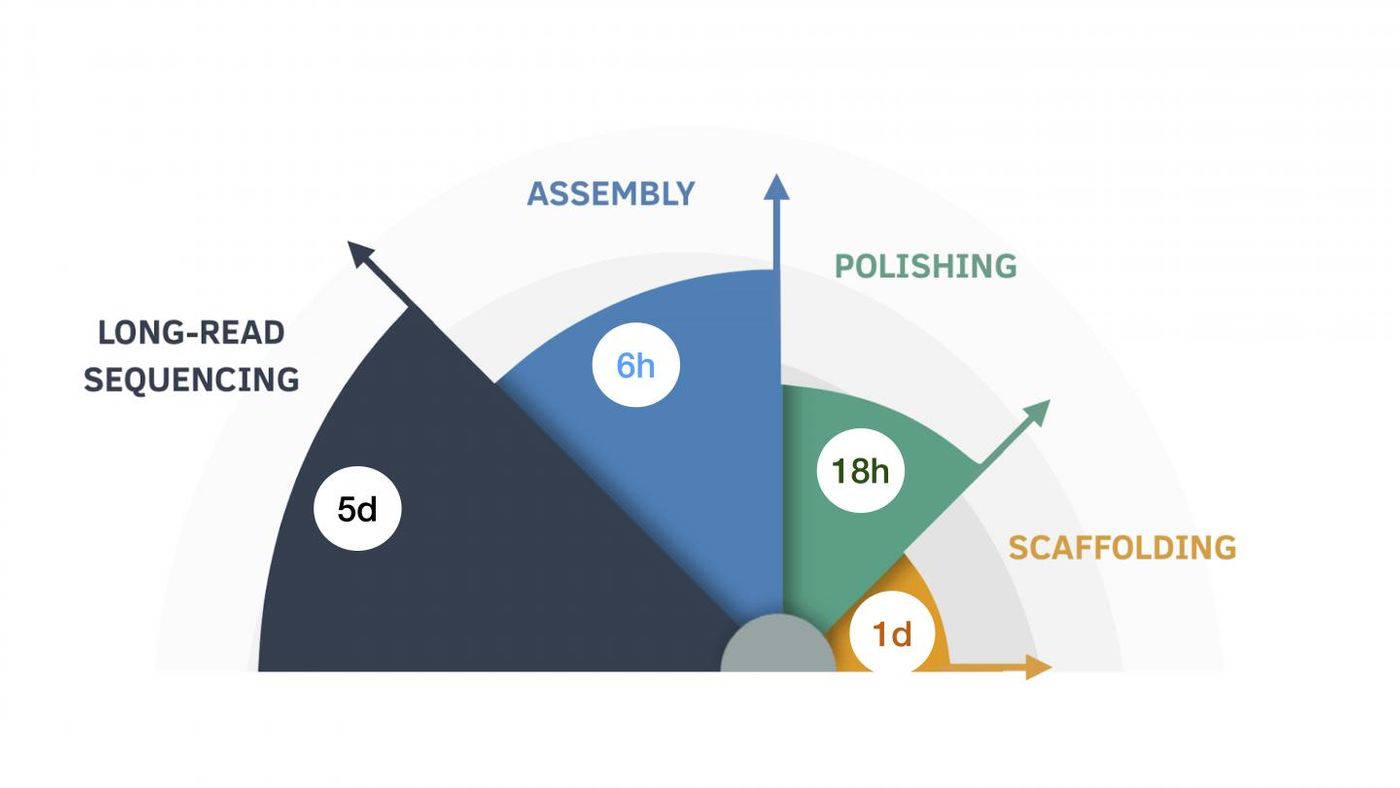
It's now been taken even further; an algorithm has been designed that can use long-read sequencing data to assemble as complete human genome in around six hours, and four about $70.
The researchers said they hope their assembler will increase the pace of genomics research and open opportunities. This includes enabling pangenome research to represent the true scale of human diversity, a decidedly more practical pursuit.
"Our new assembler was designed to be cheap and quick, with the goal to be on the cloud," said UC Santa Cruz's Benedict Paten. "It gives us the power to scale nanopore sequencing. Now, I'm confident that we'll be easily assembling hundreds of de novo genomes in the next couple of years."
Experienced research scientist and technical expert with authorships on over 30 peer-reviewed publications, traveler to over 70 countries, published photographer and internationally-exhibited painter, volunteer trained in disaster-response, CPR and DV counseling.