Researchers have created a hyper-fast, super-scalable software that shortens from weeks to mere hours the time needed to scrutinize a patient's genome for disease-causing variations.
The software is discussed in a recently published article.
"It took around 13 years and $3 billion to sequence the first human genome," says Peter White, PhD, principal investigator, Center for Microbial Pathogenesis, and director, Biomedical Genomics Core, Nationwide Children's, Columbus, Ohio, and the study's senior author. "Now, even the smallest research groups can complete genomic sequencing in a matter of days. However, once you've generated all that data, that's the point where many groups hit a wall. After a genome is sequenced, scientists are left with billions of data points to analyze before any truly useful information can be gleaned for use in research and clinical settings."
To help digest the mountain of data, White and team devised Churchill, a computational pipeline. With Churchill, scientists can deftly analyze a whole genome sample in just 90 minutes.
"Churchill fully automates the analytical process required to take raw sequence data through a series of complex and computationally intensive processes, ultimately producing a list of genetic variants ready for clinical interpretation and tertiary analysis," White says. "Each step in the process was optimized to significantly reduce analysis time, without sacrificing data integrity, resulting in an analysis method that is 100 percent reproducible."
Churchill's output has been validated with National Institute of Standards and Technology (NIST) benchmarks. White and his colleagues found its use for population-scale genomic analysis as a byproduct of its primary function. "At Nationwide Children's we have a strategic goal to introduce genomic medicine into multiple domains of pediatric research and healthcare," he says. "Rapid diagnosis of monogenic disease can be critical in newborns, so our initial focus was to create an analysis pipeline that was extremely fast, but didn't sacrifice clinical diagnostic standards of reproducibility and accuracy." Churchill is very efficient (greater than 90 percent resource utilization) and scaled adroitly across many servers.
The Churchill algorithm is licensed to GenomeNext LLC, Columbus, Ohio, a genomic informatics company, for which White is the principal genomic scientist and technical advisor.
The article, in the journal Genome Biology and titled "Churchill: an ultra-fast, deterministic, highly scalable and balanced parallelization strategy for the discovery of human genetic variation in clinical and population-scale genomics," is found here: bit.ly/1DapezP
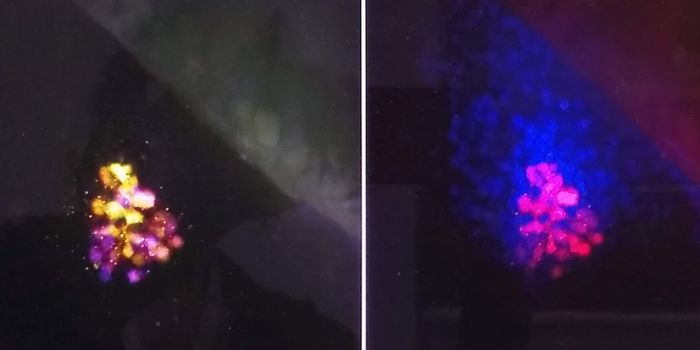
Image: Cell nucleus and chromosomes stained by Spectral karyotyping (SKY) which is a laboratory technique that allows scientists to visualize all of the human chromosomes at one time by "painting" each pair of chromosomes in a different fluorescent color. [Photo credit: Division of Intramural Research, National Human Genome Research, NIH, Bethesda, MD]