Alzheimer’s disease (AD) currently affects 5.5 million people in the U.S. over the age of 65, and that number will continue to grow unless a disease-modifying therapy is created. Even though there is a ton of research going on in the Alzheimer’s field, there are still some astounding gaps in our basic knowledge of disease pathology. It will be very difficult, if not impossible, to create the kind of disease-modifying therapy we so desperately need without a full understanding of the changes happening at the cellular level. With this in mind, a recent paper published in
Cell is very exciting because it fills in a gap in our basic Alzheimer’s disease knowledge.
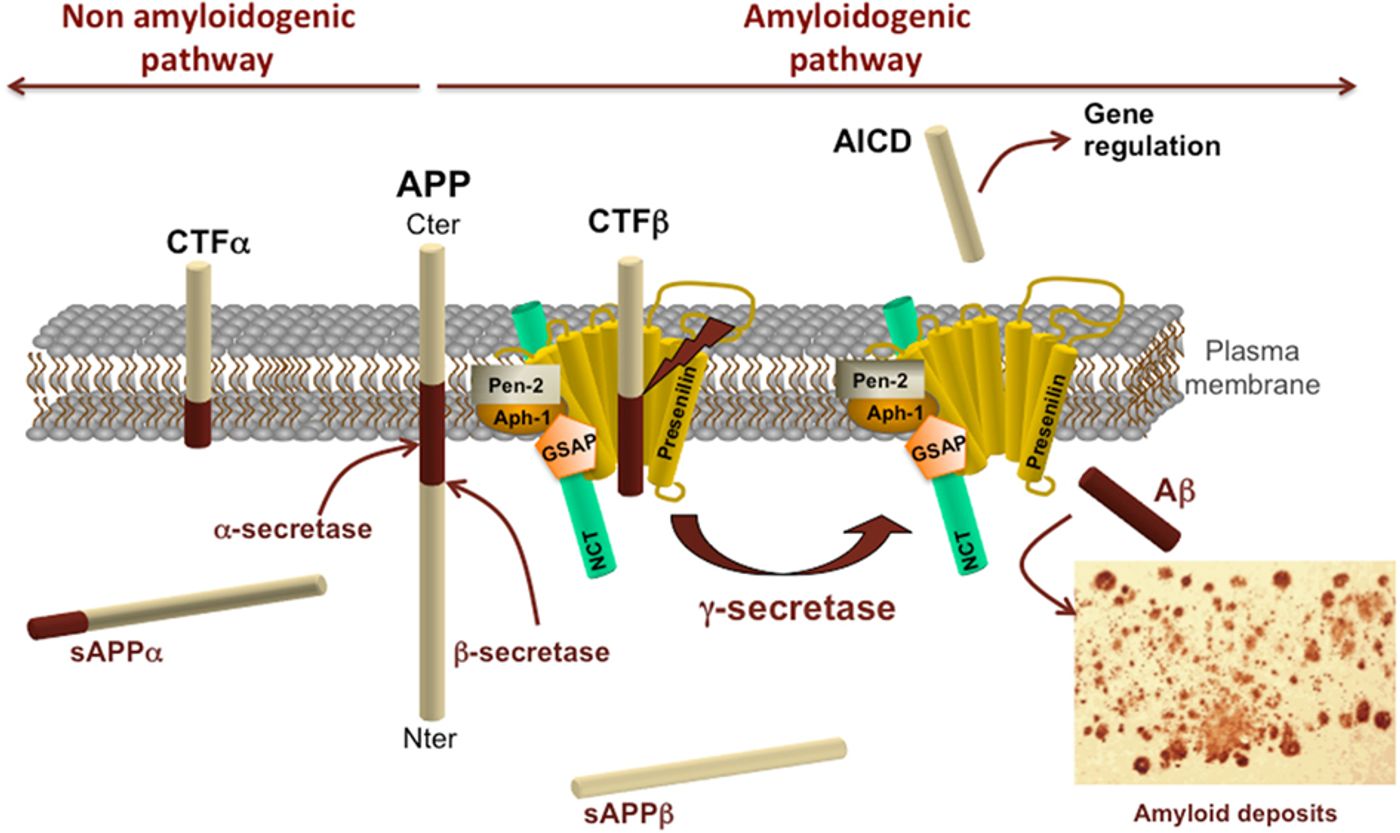
Wim Annaert’s group at KU Leuven in Belgium published a beautiful series of experiments that described the differences between presenilin 1 and presenilin 2. Presenlins are the catalytic subunit of γ-secretase, the second protease that cuts the amyloid precursor protein (APP) to form amyloid-β, the toxic peptide that accumulates in AD. Amyloid- β (Aβ) comes in a few different lengths, but for right now, we will just focus on two, Aβ40 and Aβ42. Aβ42 is considered more toxic than Aβ40 because it is more prone to aggregation, and in AD, the ratio of Aβ42:Aβ40 is much higher than in a healthy brain. There are mutations in presenilin 1 (PSEN1) that lead to higher Aβ42:Aβ40 ratios and those mutations cause familial AD. There are also some mutations in presenilin 2 (PSEN2) that lead to familial AD. Since there are only 13 known AD-causing mutations in PSEN2 compared to the over 180 known AD-causing mutations in PSEN1, most of the AD research on the γ-secretase complex has focused on PSEN1.
This new research shows that while PSEN1 is distributed among all cellular compartments, PSEN2 is localized to membranes in the endolysosomal system. The specific localization to the endolysosomal system is significant in the context of AD pathology because there is increasing evidence that endosomal dysfunction is one of the earliest pathological events in sporadic AD.
APP is cleaved by γ-secretase on endosomal membranes which generates intraneuronal Aβ. A lot of AD research has focused on extracellular Aβ in the context of plaques and synaptotoxic oligomers, but intraneuronal accumulation of Aβ plays a role in disease pathology as well. What’s more, intraneuronal pools of Aβ have a higher Aβ42:Aβ40 ratio than extracellular pools. Why exactly this is the case is unclear, but Ralph Nixon at NYU (who was unaffiliated with this study in
Cell), suggests that it might be because the aggregation-prone Aβ42 is more resistant to the acidic environment in the lysosomes so it accumulates instead of being degraded like Aβ40.
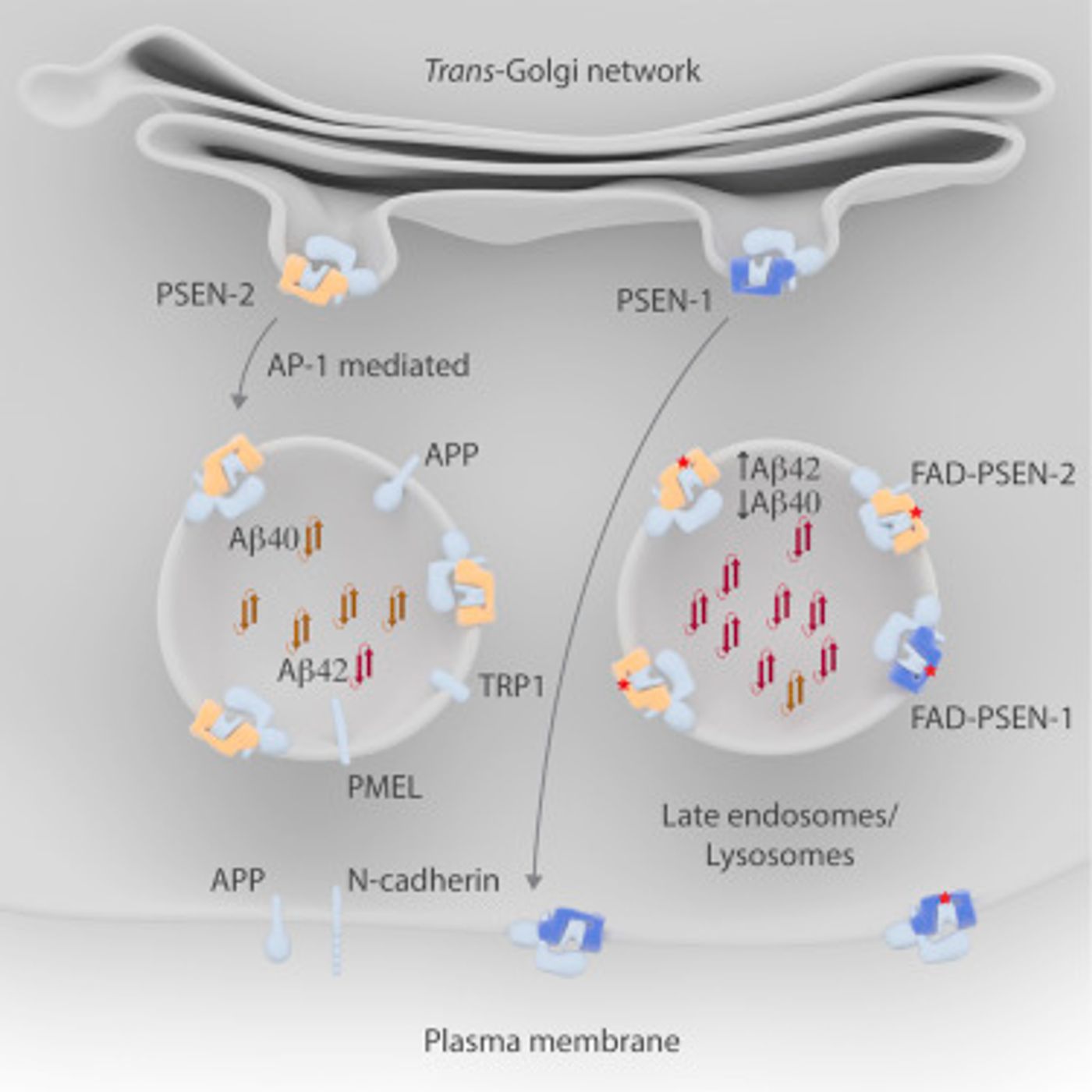
The researchers also investigated some of the known mutations in PSEN1 and PSEN2. The PSEN2 mutations did not change its localization to the endolysosomal system, but they did increase the Aβ42:Aβ40 even more. In the case of one tested mutation, the ratio of intraneuronal Aβ42 to Aβ40 was 50:1 (the normal ratio in a healthy brain is 1:9). Interestingly, they also saw that two of the known AD-causing mutations in PSEN1 that they tested changed the localization of PSEN1 so that it looked like PSEN2. The mutated PSEN1 was localized to the endolysosomal system and generated intraneuronal Aβ42. This confirms that the intraneuronal Aβ42 accumulation plays a role in AD pathology by way of a malfunctioning endolysosomal system.
This is the kind of fundamental research that we need to get a more complete picture of the early pathology in AD. If we want a disease-modifying therapy, it will most likely be most effective in the early disease stages. Understanding what is happening at this stage of disease pathology is the first step in designing a therapy to fight it.
Sources:
AlzForum,
Cell, and
Frontiers in Physiology








