Research Microbiologist/Bioinformatician, US Department of Agriculture's Dairy Forage Research Center
BIOGRAPHY
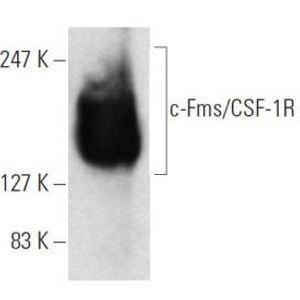
Low error rate long-reads enable the identification of long variant haplotypes in metagenome assemblies
Presented at:
Genetics Week Virtual Event Series 2021
Speaker
Abstract
Microbial communities include distinct lineages of closely related organisms which have proved challenging to separate in metagenomic assembly. Challenges include the existence of highly related organisms that exist the same environment, or several species that exist at very low abundances and are therefore inconsistently sampled. Many of these challenges are exacerbated by limitations in DNA sequencing technologies, where some platforms produce reads that are too short or reads that have too many errors. The advent of long, accurate “HiFi” reads presents a possible means to address the challenge of metagenome assembly by disambiguating individual reads or contigs within metagenomic bins. We present a metagenomic assembly of a complex microbial community from parasite-infested sheep fecal material. We found that assemblies made from HiFi reads were able to properly assemble low abundance species in our sample, and could often assemble microbial genomes into single, circular contigs. Taking advantage of the low error rates of HiFi reads, we developed a method that can track linked single nucleotide polymorphisms (SNPs), which is termed “phasing,” and identify individual SNP haplotypes that indicate subpopulations of microbes in a sample. This method is sensitive and scalable, and was able to phase haplotypes as long as 309 SNPs across hundreds of thousands of bases in the microbial genome. We also report successful characterization of multiple, closely-related microbes within a sample with potential to improve precision in assigning mobile genetic elements to candidate host genomes within complex microbial communities.
Learning Objectives:
1. Identify the challenges of metagenome assembly
2. Quantify the benefits of low error rate long reads on metagenome sequencing and assembly
3. Understand concepts such as “phasing” and “haplotyping” as it relates to metagenome assembly
You May Also Like
MAR 26, 2024 | 7:00 PM
C.E. CREDITS

The implementation of a preemptive pharmacogenomics (PGx) program in a hospital setting requires a multidisciplinary approach to ensure seamless integration of each stage of the process for...
MAR 26, 2024 | 8:00 AM
C.E. CREDITS
The implementation of a preemptive pharmacogenomics (PGx) program in a hospital setting requires a multidisciplinary approach to ensure seamless integration of each stage of the process for...
FEB 08, 2024 | 10:00 AM
High-content screening (HCS) is an imaging-based, multi-parametric strategy used in drug development that generates rich datasets through multiplexing strategically chosen fluorescent dyes a...
MAR 26, 2024 | 11:00 AM

Ever wonder what you’re missing in your data? The sheer complexity of today’s flow and mass cytometry datasets demands automated solutions. Machine learning plugins only provide...
MAR 20, 2024 | 8:00 AM
Deciphering somatic mosaicism in healthy tissues and clonal diversity in tumors necessitates single-cell analysis. High-quality genomic and transcriptomic data at the single-cell level depen...
MAR 20, 2024 | 9:00 AM
C.E. CREDITS

Join us in this enlightening session to deepen your understanding of qualifications and their transformative impact on risk management and regulatory adherence. Gain actionable insights that...
Loading Comments...
Finish Registering
-
 SEP 03, 2024Microbiology Week Virtual Event Series 2024
SEP 03, 2024Microbiology Week Virtual Event Series 2024 -
SEP 18, 2024Cell Biology Virtual Event Series 2024
-
SEP 26, 2024Olink Proteomics World
- See More






-
JUL 26, 2024
- See More