When we suffer from a cold or the flu, we usually get over the sniffling and sneezing that comes with it in time. But for some people with a genetic lung disease, primary ciliary dyskinesia, that respiratory irritation is an unfortunate way of life. They carry dysfunctional cilia, hairlike appendages that function to move mucus through our airways. They cannot efficiently push microbes out of their lungs and end up frequently getting infections. Now, researchers at Washington University School of Medicine in St. Louis have learned more about why the cilia of these patients don’t function properly.
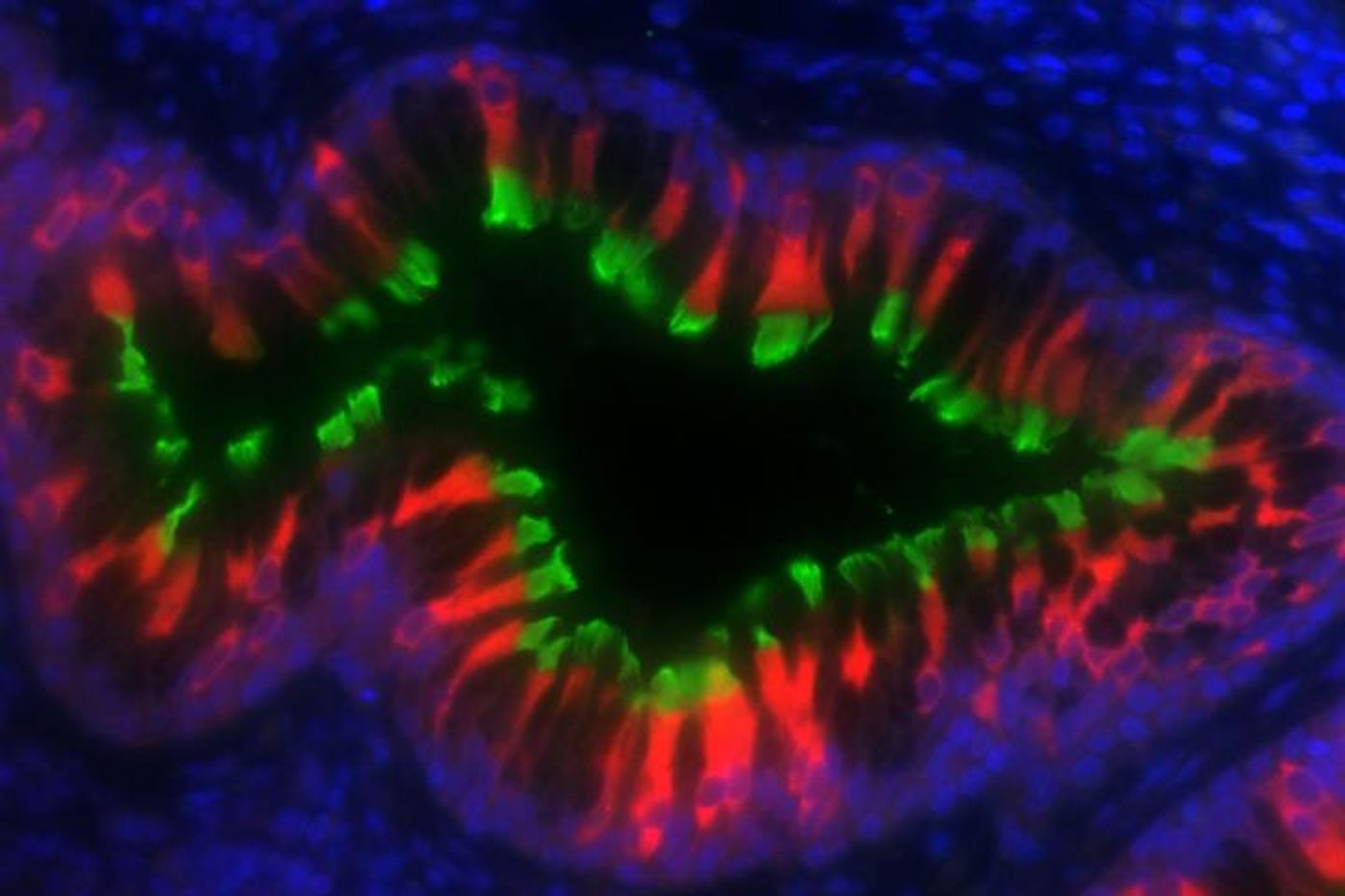
Cilia contain molecular motors that power their movement. This new work has determined that there are problems with the proteins that assemble the motors. The cilia beat is dysfunctional because nonmotor proteins don’t put the motor together properly. The study was published in the Proceedings of the National Academy of Sciences (PNAS).
"What's unique is that these failed motors actually have all their parts, and they are fully formed and normal," said senior study author Steven Brody, MD, the Dorothy R. and Hubert C. Moog Professor of Pulmonary Medicine and a professor of radiology. "It's like a machine that's missing just one screw: Everything just falls apart. If we can find a way to hold the motor together, we may be able to treat the disease. This opens a whole new opportunity to screen for medications."
Mutations in over 40 different genes have been connected to this disorder, which affects around one in 15,000 people. The disease can cause infertility as well as lung infections because the molecular motor that powers sperm and propels eggs along the female reproductive tract is the same as the one in the lungs. Only the symptoms of this disease can be treated; there is no cure.
The first author of the work, Amjad Horani, MD, is an assistant professor of pediatrics who treats around 30 children that have primary ciliary dyskinesia. He has a specialty clinic at St. Louis Children's Hospital.
Some of those patients carry mutations in the HEATR2 gene. Horani and Brody observed the HEATR2 protein in normal cells growing in culture - it was located not in cilia, but in the middle of the cells, and it was showing up very early. That pattern was similar to two other proteins that are associated with this disease.
They collaborated with David Piston, the Edward Mallinckrodt Jr. Professor and head of the Department of Cell Biology and Physiology, and staff scientist Alessandro Ustione, to show that the three proteins that have been linked to the disorder work to form a scaffold. The molecular motor is then built upon that scaffold.
The investigators followed up on their findings by growing cells that had been harvested from patients. The resulting ciliated cells showed the scaffold was indeed forming incorrectly in cells that harbored mutations in any of the genes that had been implicated. The mutated proteins can all to come together, but form a haphazard clump instead of an efficient assembly line.
"Piling together misfolded proteins turns out to be the way that the body tries to get rid of abnormal proteins," Horani explained. "Now that we know what is wrong, we want to find out, 'How can we make this work?' We're starting to study whether we can prevent the protein heaps from forming and somehow rescue the parts of the motors. If we can get just a few of these proteins to assemble, we may be able to improve quality of life for people with this disease."
Sources: AAAS/Eurekalert! Via Washington University School of Medicine, PNAS