
LA JOLLA, CA – February 2, 2017 – Chemists at The Scripps Research Institute (TSRI) have invented a new technique for constructing chiral drug molecules.
Chiral molecules are those whose structural complexity allows them to have mirror-image, “left-handed” and “right-handed” forms. For drug molecules, usually only one of those forms works—the other may even have unwanted side-effects—and thus pharmaceutical chemists have a great need for methods to build molecules in a single chiral form, rather than an even mix of both.
The new molecular construction technique, unveiled in a Science online First Release paper on February 2, 2017, represents a significant milestone in chiral chemistry. It creates a structure that chemists call an α–chiral center, thereby enabling the synthesis of a great variety of potentially valuable chiral drugs and other products. At the same time—unlike most previous chirality-inducing reactions—it requires only inexpensive and widely available starting chemicals.
“This method essentially mimics the ability of the enzymes in our cells to turn simple organic molecules into chiral molecules,” said senior author Jin-Quan Yu, Frank and Bertha Hupp Professor in the Department of Chemistry at TSRI.
Over the past decade or so, Yu and his laboratory have invented dozens of new molecule-building reactions that have been adopted widely by chemists in academia and industry. Most are C-H activation reactions, which remove a hydrogen (H) atom—the simplest and most common feature of organic molecules—from one of the carbon (C) atoms of the molecular backbone, and replace it with a more complex cluster of atoms called a functional group. Increasingly, Yu and his colleagues have designed these reactions to create the asymmetry needed for chiral drugs.
In a paper in Science last August, for example, they described a set of chiral asymmetry-making reactions that work by selectively activating just one of the two hydrogen atoms on a methylene group (CH2), a feature of many organic molecules. Like most of the C-H activations developed by the Yu Laboratory, this reaction employs a palladium atom to break the targeted C-H bond, and a special “ligand” molecule to steer the palladium atom precisely where it needs to go.
The new set of reactions has an even more challenging target: a cluster of carbon and hydrogen atoms known as an isopropyl group, another feature of many organic molecules. The ligands developed for the reaction, derivatives of aminomethyl oxazoline, effectively select a carbon at just one side of the isopropyl group and replace one of its hydrogen atoms with a functional group.
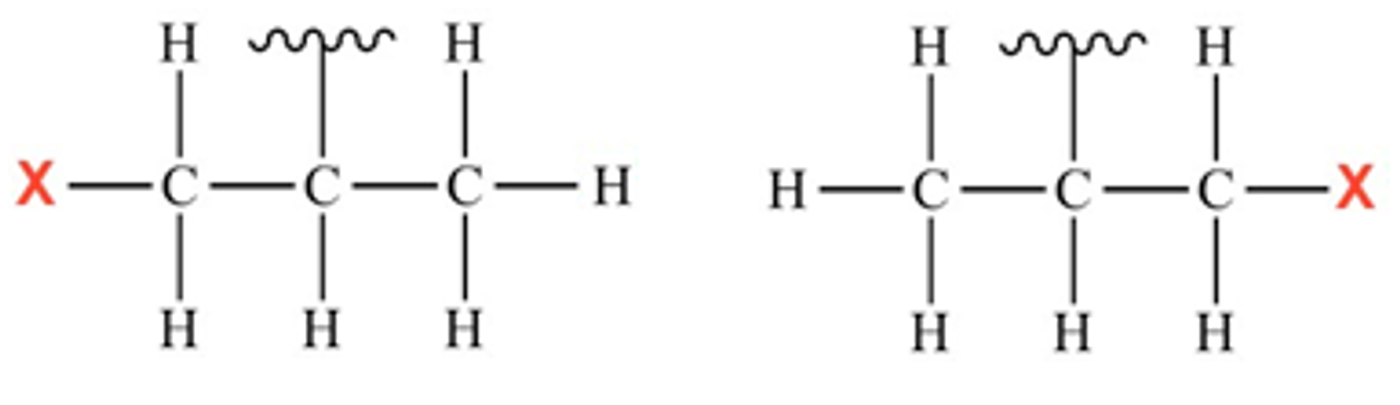
Isopropyl group with functional group attached on one side or the other
Yu’s team showed that they can use the ligands to add aryl, alkene and alkyne functional groups—common building-blocks in the construction of drug molecules.
The intended starting material for the new reactions is an isopropyl-bearing molecule called isobutyric acid, although the reactions also work well on related molecules. Isobutyric acid is cheaply produced in very large quantities using standard industrial methods, and it can also can be generated from waste biomass such as crushed sugar cane—making it a more environmentally friendly ingredient for chemical reactions.
Isobutyric acid is also found in nature, and bacteria and other organisms have evolved enzymes that convert it to natural chiral molecules. In the past few decades, pharmaceutical chemists have learned to harness some of these natural enzyme reactions—using genetically engineered bacteria—to help them build chiral drug molecules. However, these enzyme-driven reactions are restricted to isobutyric acid as a starting molecule and are very limited in the chiral molecules they can yield. The new ligand/catalyst, while essentially mimicking nature’s synthetic feat, is much more versatile.
“Now that we know how to selectively break that one C-H bond with a palladium catalyst, we’re not limited to the reactions that enzymes can do,” Yu said.
He added that researchers at Bristol-Myers Squibb, which has a research collaboration agreement with the Yu Laboratory, are already using the new reactions to make potential new drug molecules.
The first author of the paper, “Formation of α-chiral centers by asymmetric β-C(sp3)–H arylation, alkenylation, and alkynylation,” was Qing-Feng Wu, a postdoctoral research associate in the Yu Laboratory. The other co-authors were Claudio Mapelli, Jennifer Qiao and Michael Poss of Bristol-Myers Squibb; and Peng-Xiang Shen, Jian He, Xiao-Bing Wang, Forrest Zhang and Qian Shao of TSRI.
Support was provided by the National Institutes of Health (grant 2R01GM084019) and Bristol-Myers Squibb.
This article was originally published on scripps.edu.








