Scientists have now characterized a genetic disorder called linkage-specific-deubiquitylation-deficiency-induced embryonic defects syndrome or LINKED; it's caused by mutations in the OTUD5 gene and causes developmental delays and brain, facial, and heart defects. The research suggested that the mutated OTUD5 protein may be involved in a critical developmental pathway that underlies other developmental disorders.
"Our discovery of the dysregulated neurodevelopmental pathway that underlies LINKED syndrome was only possible through the teamwork of geneticists, developmental biologists, and biochemists from NIH," noted lead study author Achim Werner, Ph.D., an investigator at the National Institute of Dental and Craniofacial Research (NIDCR). "This collaboration provided the opportunity to pinpoint the likely genetic cause of disease, and then take it a step further to precisely define the sequence of cellular events that are disrupted to cause the disease."
This study began as many have: with a patient. That individual had severe birth defects that impacted the brain, heart, craniofacial skeleton, and urinary tract. After studying the patient's genome and that of their family members with genetic and computational tools, the OTUD5 mutation was identified as the likely cause of the disorder. After reaching out to other researchers in search of similar patients that were willing to volunteer their genetic material for the study, the researchers found several other OTUD5 mutations in males ranging from one to fourteen years of age.
The gene encodes for an enzyme, also called OTUD5, which helps modify proteins in a process called ubiquitylation.
"Based on the genetic evidence, I was pretty sure OTUD5 mutations caused the disease, but I didn't understand how this enzyme, when mutated, led to the symptoms seen in our patients," said co-first study author David B. Beck, M.D., Ph.D., a clinical fellow at the National Human Genome Research Institute (NHGRI). "For this reason, we sought to work with Dr. Werner's group, which specializes in using biochemistry to understand the functions of enzymes like OTUD5."
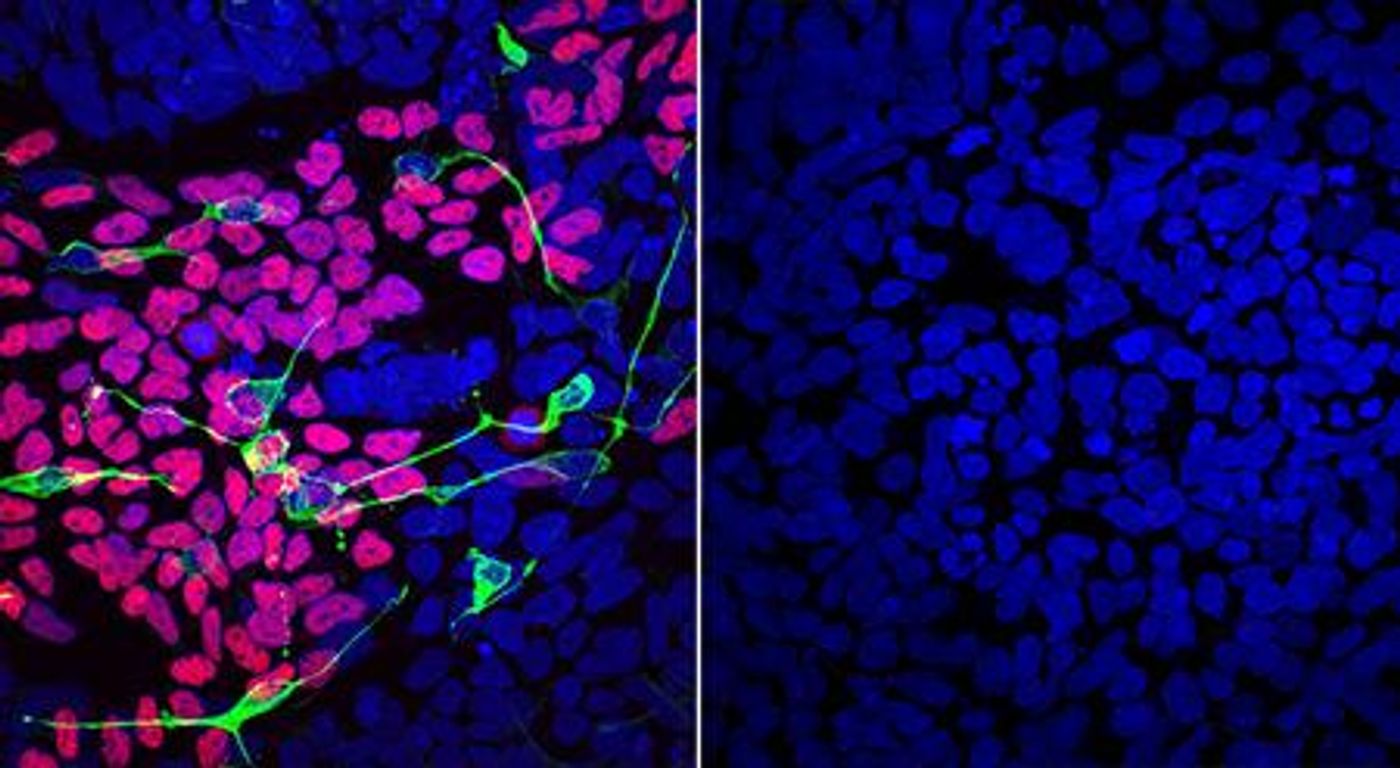
The researchers were also able to study cells collected from patients. By comparing them to cells from healthy people, the scientists found that OTUD5 would normally edit molecular tags on some proteins to control their function. But when OTUD5 was mutated, it couldn't perform its duty.
Further work showed that OTUD5 normally prevents chromatin remodelers from being marked for destruction. But when mutant OTUD5 can no longer protect these chromatin remodelers, they're destroyed. That leads to the abnormal development of neural precursor and neural crest cells, which are crucial to development.
"Several of the chromatin remodelers OTUD5 interacts with are mutated in Coffin Siris and Cornelia de Lange syndromes, which have clinically overlapping features with LINKED syndrome," noted Werner. "This suggests that the mechanism we discovered is part of a common developmental pathway that, when mutated at various points, will lead to a spectrum of disease."
"We were surprised to find that OTUD5 elicits its effects through multiple, functionally related substrates, which reveals a new principle of cellular signaling during early embryonic development," said co-first study author Mohammed A. Basar, Ph.D., a postdoctoral fellow in Werner's lab. "These findings lead us to believe that OTUD5 may have far-reaching effects beyond those identified in LINKED patients."
"We're finally able to provide families with a diagnosis, bringing an end to what is often a long and exhausting search for answers," added Beck.
Experienced research scientist and technical expert with authorships on over 30 peer-reviewed publications, traveler to over 70 countries, published photographer and internationally-exhibited painter, volunteer trained in disaster-response, CPR and DV counseling.