Clinical & Molecular DX
A Crystal Ball for Predicting Labor Day
MAY 18, 2021 7:00 AM PDT
Share
Library Prep Methods for SARS-CoV-2 Sequencing: a Summary
As the SARS-CoV-2 virus continues to mutate, rapid sequencing of COVID-19-positive samples is more critical than ever. Next-generation sequencing (NGS) is the best technology for identifying both known and emerging variants, including those containing multiple mutations; the detailed data provided by NGS will aid in understanding how these variants impact the effectiveness of diagnostic tests, vaccines, and therapies.
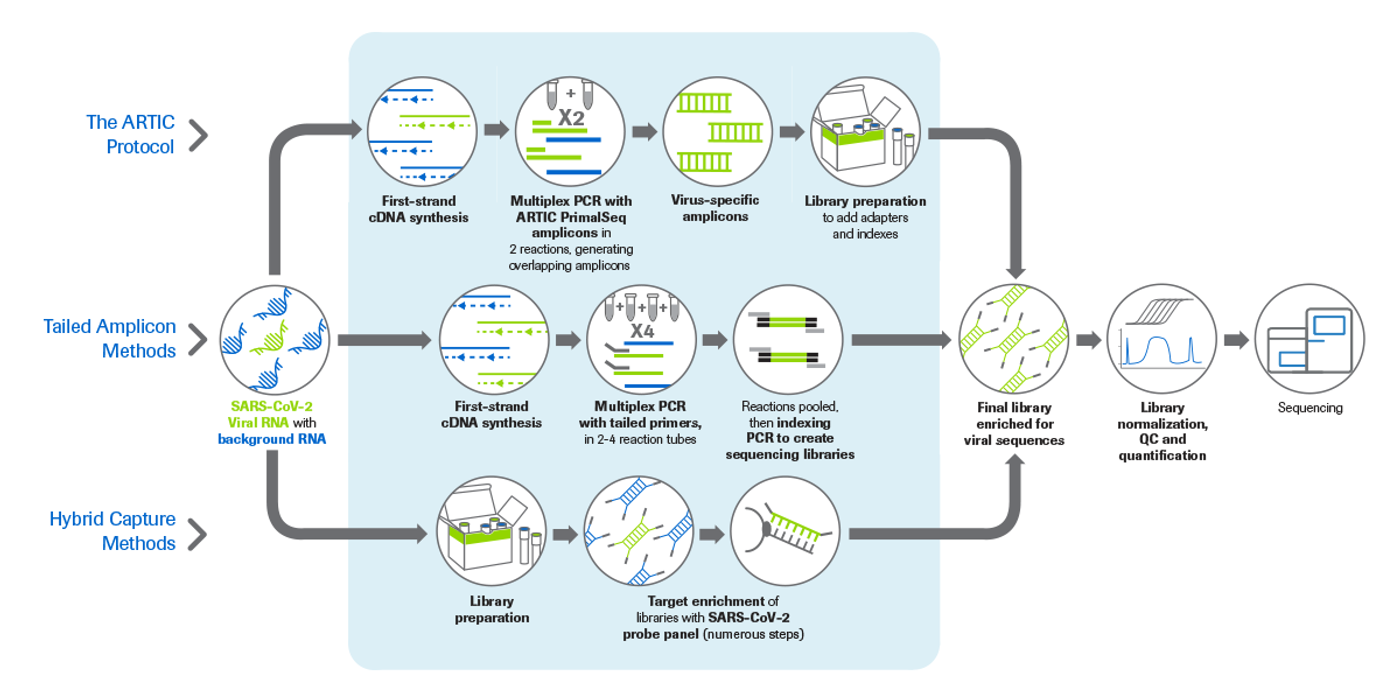
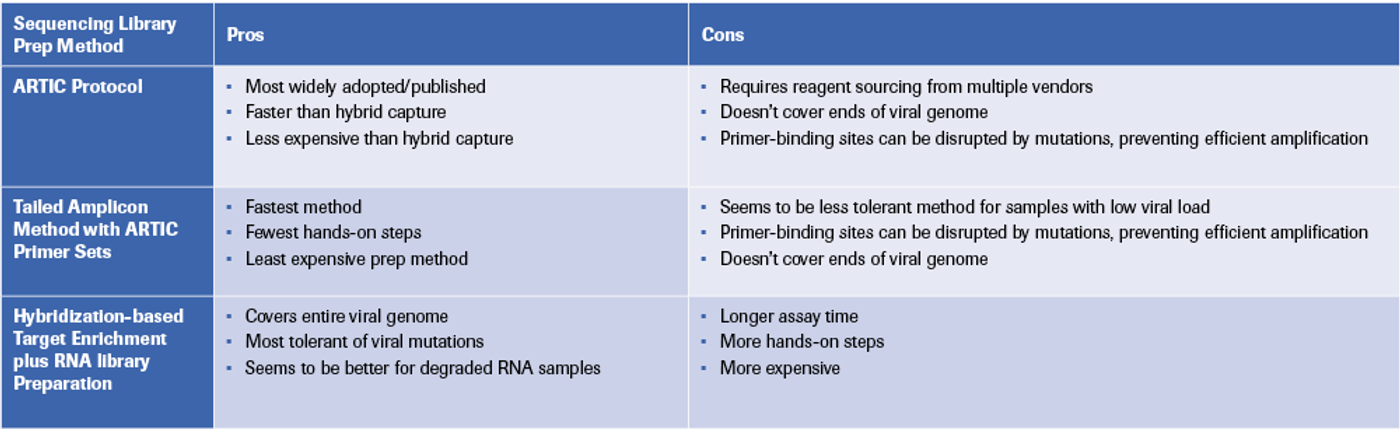
To meet this urgent need, several methods have been developed for sequencing the viral genome from RNA samples containing both human and viral RNA. Here, we summarize three approaches for generating enriched SARS-CoV-2 sequencing libraries (Figure 1) and provide insights into the strengths and limitations of each (Table 1).
The ARTIC Protocol
The ARTIC protocol is the most widely published method. This method uses publicly available primer sequences, and pooled primers can be ordered from oligo manufacturers. It is very effective with high-quality RNA, and it costs less than some other methods.
This ARTIC protocol is based on the PrimalSeq technique first developed for sequencing the Zika virus and was created by the UK-based ARTIC Network. In this method, cDNA is first synthesized from the RNA sample, then combined with two SARS-CoV2-specific ARTIC primer pools in multiplexed PCR reactions to generate overlapping amplicons across the viral genome. The resulting amplicon pools are then combined and used as input for conventional NGS library preparation. Disadvantages include the inability to detect variants outside of the amplicons (e.g., at the genome termini) and the potential disruption of primer-binding sites by new viral mutations.
Tailed Amplicon Methods
The Tailed Amplicon method is another amplicon-based option for generating SARS-CoV2 libraries. This method is fast, cost-effective, and works well with high-quality RNA.
Like the ARTIC protocol, this workflow starts with cDNA synthesis, followed by cDNA amplification via multiplexed PCR to enrich the viral genome. Here, however, the ARTIC primers are modified to contain additional adapter tail sequences, which enable the addition of indexed adapters in a second PCR. This “indexing PCR” replaces the multistep library prep used in the ARTIC protocol. While this reduces the number of overall steps, the tailed sequences on the primers require that the PCR be carried out in four reactions—rather than two— increasing the number of tubes.
Libraries created with this method suffer from the same limitations described above. In addition, this method requires the user to order and pool ~98 individual primers and is less effective than the ARTIC protocol with low-viral-load samples.
Hybrid Capture Methods
Another alternative is hybridization-based enrichment of the SARS-CoV-2 genome using oligonucleotide probes. Although more complex, these methods can detect variants across the whole genome, including genome termini. They are also more tolerant of mutations in regions that could prevent binding by ARTIC primers, and they are better suited for degraded RNA samples (e.g., wastewater).
These workflows often start with an RNA library preparation kit to perform cDNA synthesis and generate sequencing libraries from RNA. The libraries are then combined with biotin-labeled SARS-CoV-2-specific oligonucleotide probes that bind to library molecules containing viral sequences. The labeled, enriched libraries are then purified and sequenced.
Despite the benefits of hybrid capture methods, they are more expensive, have more hands-on steps, and take longer than amplicon-based methods, although progress is being made in reducing the length and complexity of these workflows.
Choosing a Workflow
When choosing the best method for your lab it is important to consider several factors, including the available sequencing platforms, experimental goals, required turnaround time, budget, and sample quality. In addition, multiple methods may be required as surveillance needs, research goals, and the virus continue to evolve.

You May Also Like
SEP 02, 2025
Genetics & Genomics

The domestication of horses is a much more recent even compared to other domesticated species; horses were domesticated
...
NOV 10, 2025
Genetics & Genomics
The 2024 Nobel Prize in Physiology or Medicine was awarded to Victor Ambros and Gary Ruvkun for their groundbreaking dis
...
OCT 20, 2025
Cell & Molecular Biology

Mitochondria are incredible organelles. They not only provide crucial power to our cells, these little machines also hav
...
NOV 26, 2025
Microbiology
Bacterial cells can easily swap bits of DNA, and many of those pieces of genetic material carry genes that confer resist
...
JAN 07, 2026
Genetics & Genomics

There is a genetic feature known as a repeat expansion, in which short sequences are repeated continuously. Some of them
...
JAN 12, 2026
Genetics & Genomics

In patients with retinitis pigmentosa (RP), crucial cells in the retina known as rods and cones die over time; night bli
...
Loading Comments...
