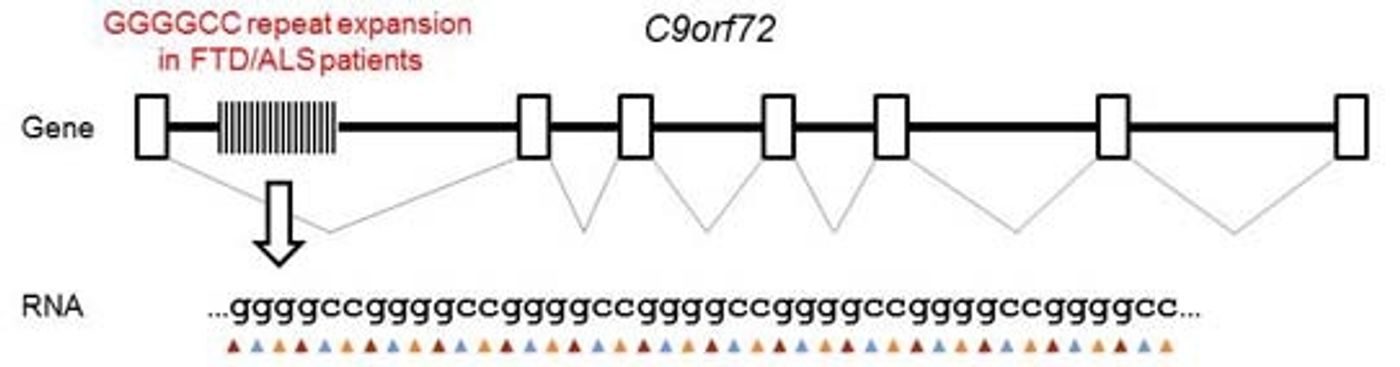
Alzheimer’s disease gets most of the press for neurodegeneration, but it is not the only devastating neurodegenerative disease out there. Frontotemporal dementia (FTD) and amyotrophic lateral sclerosis (ALS) are both less prevalent than Alzheimer’s, but they present their own issues and challenges. ALS affects ~30,000 people in the U.S. and is relatively fast-moving, with an average life expectancy from time of diagnosis of 2-5 years. FTD is slightly more prevalent, affecting ~50,000-60,000 in the U.S. What is interesting about FTD is that the average age of diagnosis is 57, a full 13 years earlier than the average age of Alzheimer’s diagnosis. ALS is characterized by the degeneration of motor neurons and FTD is more cognitive and behavioral deficits, but there is some evidence that these two diseases share an underlying pathology, specifically the G
4C
2 repeat expansions in
C9ORF72.
ALS and FTD that are caused by these repeat expansions are collectively referred to as c9FTD/ALS. This repeat expansion can be transcribed into sense and antisense mRNA which is then translated to produce a series of proteins of repeating dipeptides. Using the single letter code for amino acids, these dipeptides are GP, GA, GR, PA, and PR. Researchers at the Mayo Clinic Department of Neuroscience were especially interested in the role the poly(GA) peptides play in disease pathogenesis.
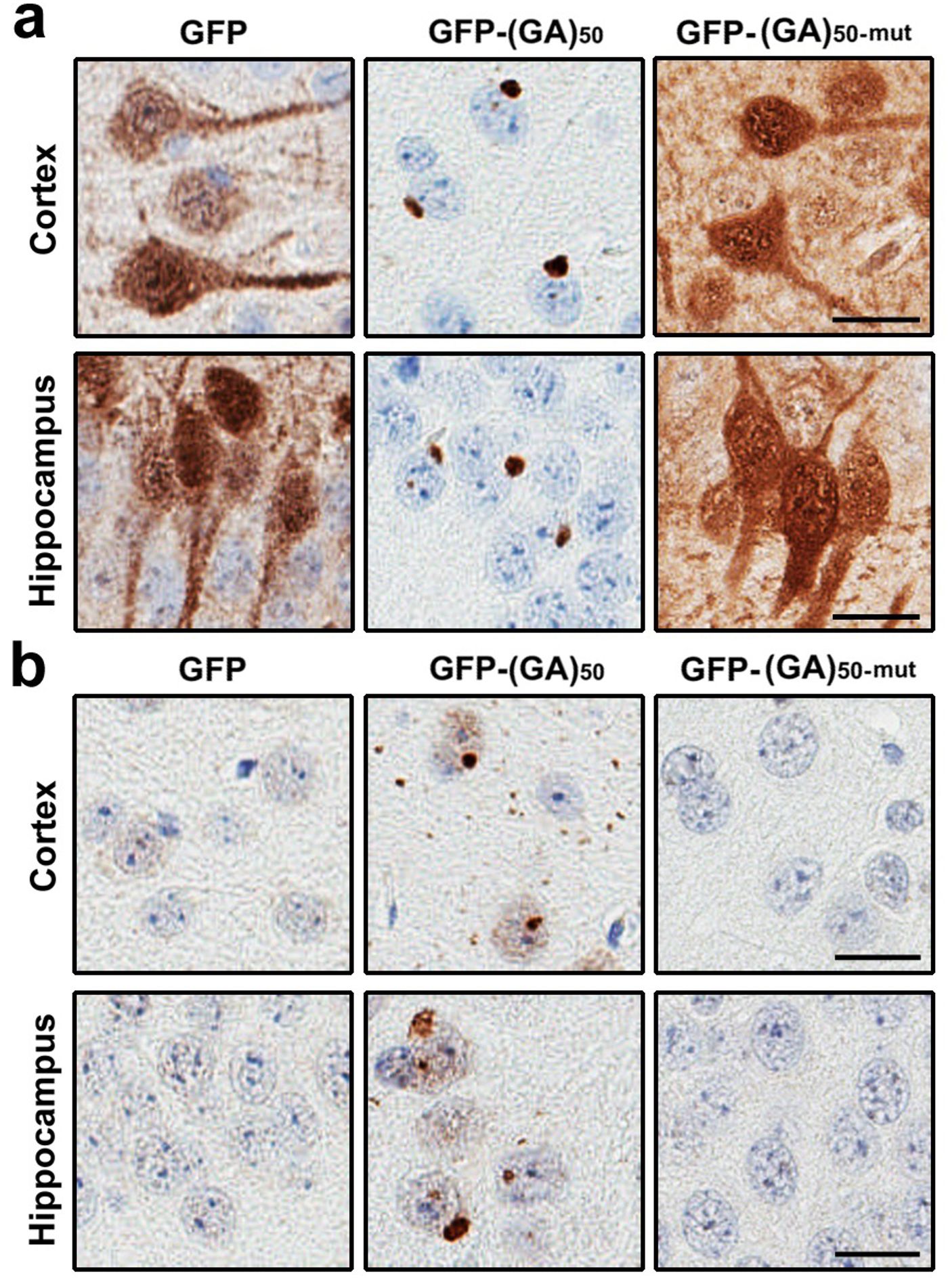
In the brains of patients with c9FTD/ALS, there are abundant poly(GA) inclusions in the cytoplasm of neurons. Applying poly(GA) to cultured cells or primary neurons induced toxicity, lending validity to their theory that poly(GA) aggregates have a strong contribution in c9FTD/ALS disease pathology. To study the mechanisms behind poly(GA)’s pathological role in c9FTD/ALS, the researchers used a mouse model that specifically looks at poly(GA) with no other pathologies. To create this mouse model, they used an adeno-associated viral (AAV) vector to express poly(GA) in the mouse central nervous system. They also wanted to specifically look at the pathological role of aggregated poly(GA), so they created a poly(GA) mutant [poly(GA)
mut] that is incapable of aggregating. The poly(GA)-expressing mice had a very strong neurodegeneration phenotype, with abundant poly(GA) inclusions; neuronal loss in the cortex, hippocampus, and cerebellum; and behavioral, motor, and memory deficits. The poly(GA)
mut mice had none of these signs of neurodegeneration, suggesting that poly(GA) pathology is dependent on poly(GA) aggregation.
The most important finding from this paper though is the discovery that poly(GA) aggregation leads to HR23 pathology. HR23A and HR23B are proteins that are involved in DNA repair and in shuttling proteins that are marked for degradation with ubiquitin to the proteasome. The researchers saw that ubiquitinated proteins and HR23 proteins accumulate in poly(GA) inclusions. The model they propose is that poly(GA) aggregates sequester HR23 proteins, leading to an accumulation of ubiquitinated proteins in the cytoplasm. Have a build-up of proteins that need to be degraded is very cytotoxic.
This paper is extraordinarily detailed and I have barely scratched the surface of the experiments that the researchers did. The main takeaway is that poly(GA) aggregation contributes significantly to c9FTD/ALS neurodegeneration by initiating HR23 pathology and impairing the ubiquitin-proteasome system. It also hints at the possibility of targeting the poly(GA) monomers to inhibit their aggregation as a therapeutic strategy. That kind of therapy is a long way away, but research like this is the first step.
Sources:
Nature Neuroscience,
ALS, and
FTD