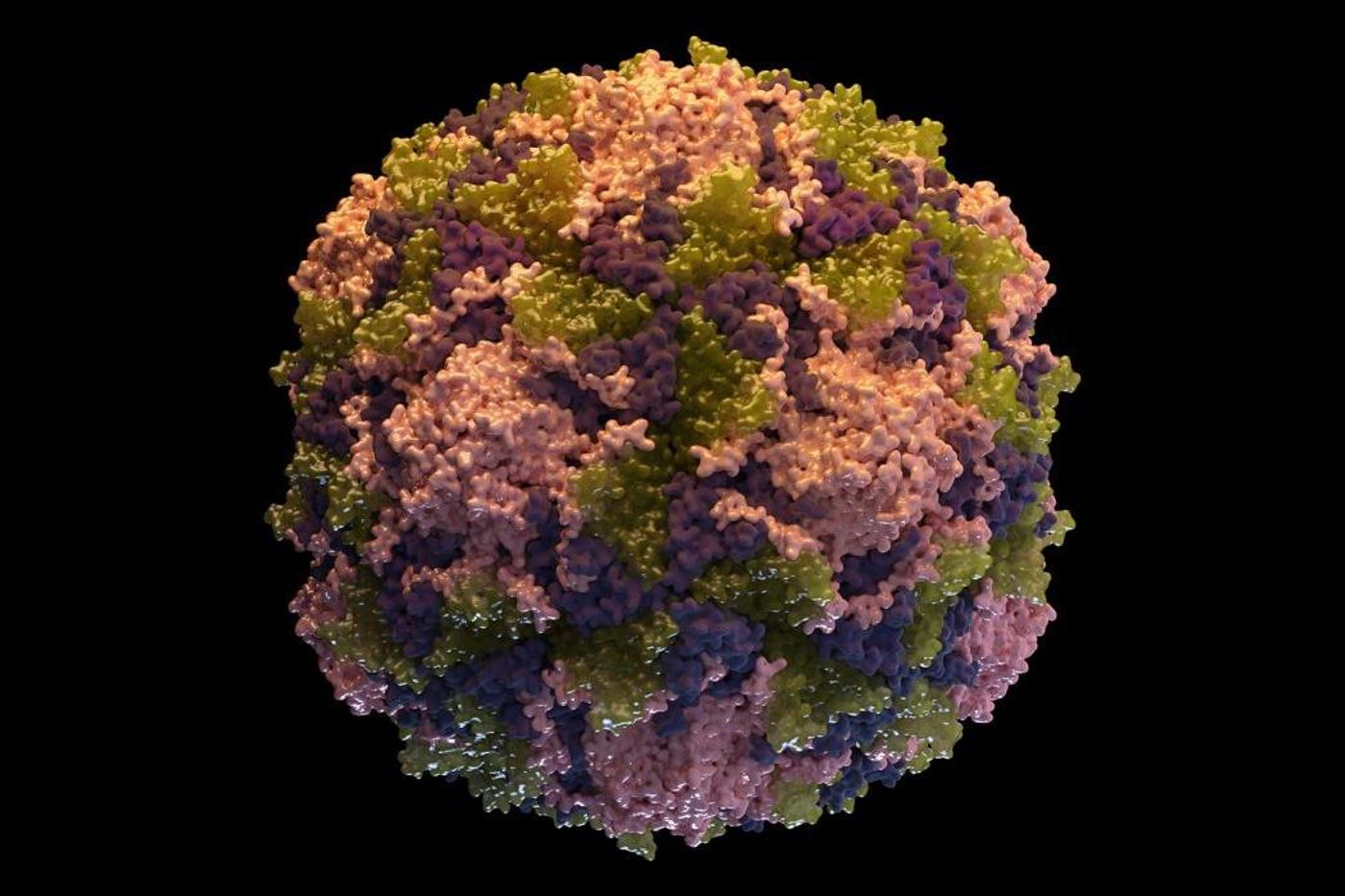
Colds don’t usually last very long or cause serious illness, but they’re annoying and extremely common. They’re caused by rhinoviruses, and there are about 160 different kinds of them that we know of. The rhinovirus genome can mutate or change rapidly, making them very difficult to fight with our immune system, a drug, or a vaccine. Now scientists at the University of California-San Francisco and Stanford have reported in Nature Microbiology that temporarily disabling a protein in cells caused a broad range of viruses, including rhinoviruses and enteroviruses, to stop replicating inside of both cells that grew in culture and a mouse model.
"Our grandmas have always been asking us, 'If you're so smart, why haven't you come up with a cure for the common cold?'" said study author Jan Carette, Ph.D., associate professor of microbiology and immunology. "Now we have a new way to do that."
Many viruses that have been well-studied carry very small genomes, and need the cellular machinery that host cells can provide in order to replicate, spread more viral particles, and infect more cells. By targeting proteins in the cells that host the virus, the researchers disrupted viruses that are linked to brain and heart inflammation, asthma, and polio - poliovirus is one type of enterovirus.
In this work, the researchers genetically-engineered cells to randomly disabled nearly every gene, one at a time, with gene editing. They infected these engineered human cells with a rhinovirus that makes asthma worse, called RV-C15, and EV-C68, an enterovirus linked to acute flaccid myelitis. The viruses use different host proteins to thrive. Some cells made it through the infection and generated offspring, and the researchers determined which genes were deleted in those cells. There were also a few cells that survived both infections, and the researchers zeroed in on one gene disabled in these cells, SETD3.
"[SETD3] was clearly essential to viral success, but not much was known about it," Carette explained.
When the researchers used other types of viruses, including other types of rhinovirus and two types of coxsackievirus to infect SETD3-deficient human cells, none were able to replicate. Compared to cells with normal SETD3 expression, viral replication was reduced in the SETD3-deficient cells 1,000-fold when infected with rhinovirus and about 100-fold when infected with an enterovirus.
Mice that were engineered to lack SETD3 appeared to live normally to adulthood and were fertile, but they could not be infected with two different enteroviruses - even when the viruses were injected directly into the rodent’s brains just after birth.
"In contrast to normal mice, the SETD3-deficient mice were completely unaffected by the virus," Carette said. "It was the virus that was dead in the water, not the mouse."
The researchers determined that enteroviruses don’t use the enzymatic function of SETD3 that cells need normally. Enteroviruses have a protein that interacts with another different part of the SETD3 protein, and that is required in some way for the virus to replicate.
"This gives us hope that we can develop a drug with broad antiviral activity against not only the common cold but maybe all enteroviruses, without even disturbing SETD3's regular function in our cells," Carette added.
Experienced research scientist and technical expert with authorships on over 30 peer-reviewed publications, traveler to over 70 countries, published photographer and internationally-exhibited painter, volunteer trained in disaster-response, CPR and DV counseling.