For people that suffer from diseases with no treatments, the outlook can be bleak. One such illness is Charcot-Marie-Tooth disease, a hereditary disorder that while relatively rare, is the most common inherited neurological disease, affecting around 3 million people worldwide. Its hallmark is a gradual loss of motor neurons that eventually leads to paralysis. Watch the video bellow to learn more about the disease.
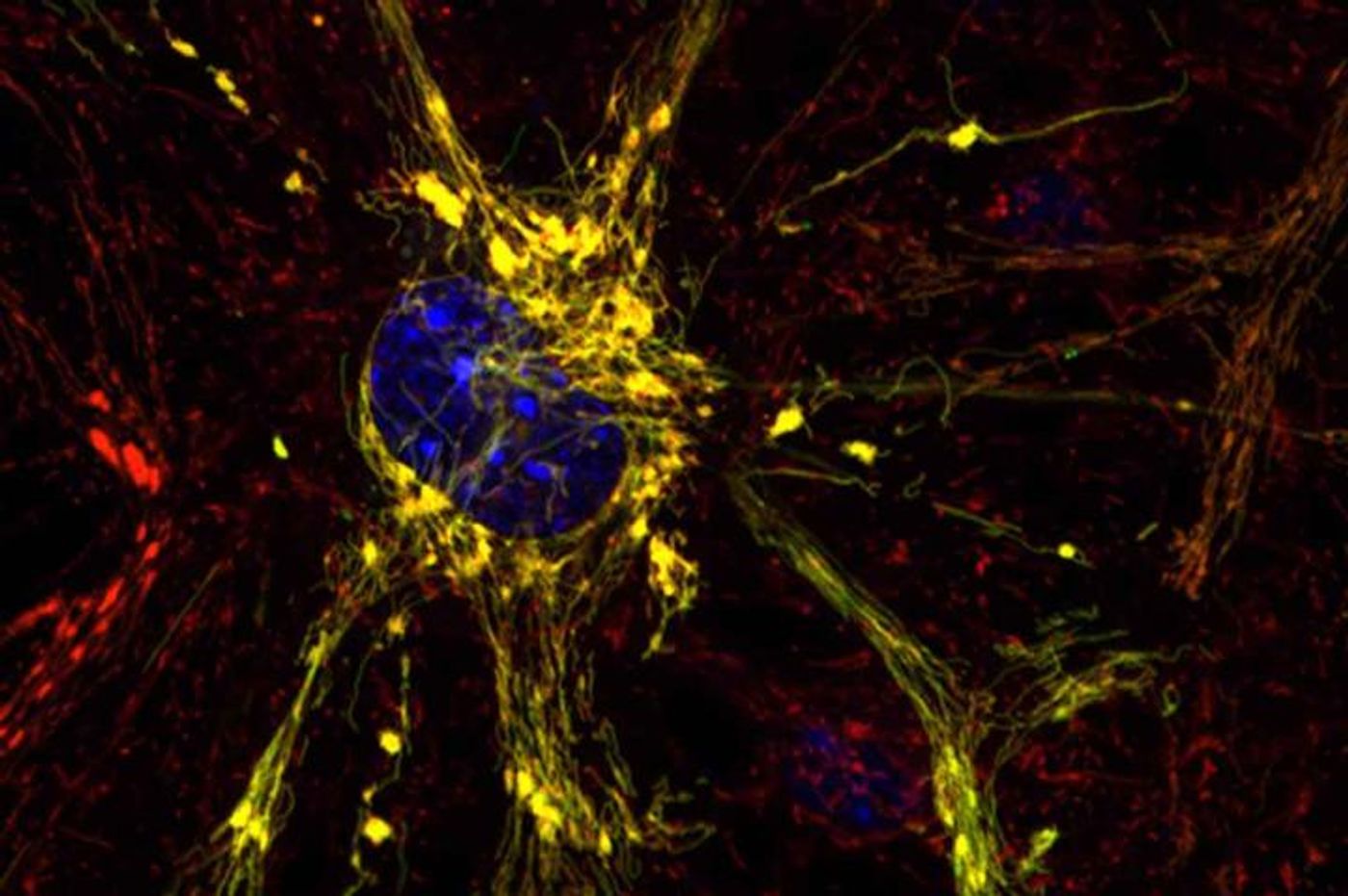
In new work published today in Nature, researchers from the Washington University School of Medicine in St. Louis and Stanford University have reported that small molecules of their design could potentially help to correct the cellular dysfunction in mitochondria that causes Charcot-Marie-Tooth and other diseases of that organelle. Analysis of mouse cells led the investigators to a new understanding of the three-dimensional structure of a crucial protein that is not functional in the mitochondria of those with the disease.
"This mitochondrial protein has never been targeted before," explained the senior author of the work, Gerald W. Dorn II, MD, the Philip and Sima K. Needleman Professor of Medicine. "There are no drugs that work on this protein that is so important for mitochondrial function. We designed two compounds -- one that activates and one that inhibits the function of this protein. We are working on testing them in mice with mitochondrial defects."
The protein the scientists investigated is called mitofusin 2, and it could play a role in a wide variety of disorders including heart disease and diabetes. While those diseases are not thought of as mitochondrial in nature, mitofusin 2 controls the interaction between two mitochondria, controlling whether they bind one another and fuse to exchange genetic information. That process is thought to be vital to the health of mitochondria, which is critical to the health of cells and tissues.
"In the past, scientists assumed mitofusin 2 was always active, always ready to tether to another mitofusin molecule and promote mitochondrial fusion," said Dorn. "Our study now shows this is incorrect. Mitofusin 2 folds and unfolds, giving it active and inactive forms that either encourage or discourage tethering and the resulting fusion of mitochondria."
After Dorn’s team revealed the mechanism underlying the conformational changes of the protein, they could then design peptides that would interact with the protein, moving it toward an active or an inactive state.
"We designed these molecules based on our new knowledge of mitofusin 2," Dorn commented. "My colleague, Dr. Mochly-Rosen, is a genius at designing this kind of small peptide drug. She looks at amino acid sequences and sees things I don't see."
One such molecule is called GoFuse. It drives mitofusin 2 to the healthy, active state and by extension, encourages the tethering and fusing of mitochondria. Another molecule, called TetherX, pushed mitofusin 2 into an inactive state, which halts tethering and thus prevents fusing.
"The design of these peptide inhibitors was a challenge," Mochly-Rosen said. "But it is always exciting when a basic research discovery leads to the design of a new drug that may eventually help patients who currently have no treatment options."
More work remains to evaluate how these peptides will work in animal models. The aim is that GoFuse or a molecule like it will cause the behavior that is missing in Charcot-Marie-Tooth. The ultimate goal is to prevent the motor neuron loss in the disease, and halt the paralysis seen in patients. The researchers also hope that their work will have an impact beyond just this disease as well. One potential is the reduction of tissue damage seen in patients that have suffered a stroke.
"Re-establishing oxygen flow is really important after a heart attack or stroke," Dorn explained. "But you also get a huge wave of cell death when oxygen suddenly returns to tissues of the body, such as the heart or the brain."
As oxygen rushes back into tissues, an influx of calcium occurs in mitochondria that are tethered. The massive amount of calcium that flows into mitochondria also causes water to rush in; all that extra material overflows the mitochondria, which burst, killing the cell. Dorn speculated that if that kind of tethering was suppressed, it might prevent the huge influx of calcium, thus protecting mitochondria from being destroyed.
"These peptides are two sides of the same coin," Dorn continued. "Mutations that disrupt tethering cause a neurodegenerative disease. We would like to encourage tethering in that case. But there are other situations where tethering is destructive, and we would like the ability to interrupt it briefly and then go back to normal. We've shown these peptides can influence mitochondrial tethering in cells grown in the lab, and now we are working to test them in mouse models of disease."
Sources:
AAAS/Eurekalert! via
Washington University School of Medicine,
Nature