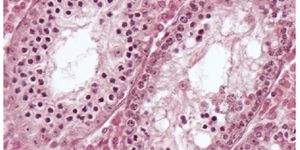
In an advance that could lead to better identification of malignant pediatric adrenocortical tumors, and ultimately to better treatment, researchers have mapped the "genomic landscape" of these rare childhood tumors. Their genomic mapping has revealed unprecedented details, not only of the aberrant genetic and chromosomal changes that drive the cancer, but the sequence of those changes that trigger it.

The study was led by Raul Ribeiro, M.D., Jinghui Zhang, Ph.D., and Gerard Zambetti, Ph.D., all members respectively, of the Departments of Oncology, Computational Biology and Pathology at St. Jude Children's Research Hospital. The researchers published their findings online in the journal Nature Communications. First authors of the paper were Emilia Pinto, Ph.D., St. Jude Pathology, and Xiang Chen, Ph.D., St. Jude Computational Biology.
Understanding the genetic machinery that drives these tumors is critical because of the difficulty in reliably classifying which childhood adrenocortical tumors would prove to be malignant. Currently, only about half of children with these tumors remain cancer-free after treatment, and those with advanced cancers have very poor overall survival.
"We haven't had any good markers to establish a prognosis," Ribeiro said. "The only characteristic that was somewhat consistent was tumor size, with larger tumors having a worse outcome than smaller ones. But even then, we would have cases where patients with large tumors would have good prognoses, and those with smaller tumors would do poorly."
Zambetti said scientists in the field had identified only a few genetic markers that seemed to predispose children to these tumors.
"Pediatric adrenocortical tumors had never been analyzed on a genomic scale before," Zambetti said. When the researchers sequenced the genomes of the tumor and blood samples from the 37 patients with early-to late-stage disease, they pinpointed key mutations involved in these tumors as well as their timing in cancer development.
One key genetic mutation was detected in the gene called TP53, which resides on chromosome 17p. TP53 acts as a "brake" on cell division under stress conditions, so its inactivation by mutation would unleash the uncontrolled proliferation of cancer cells.
A second key molecular event uncovered by the study occurs on chromosome 11. This chromosome harbors a gene called IGF2, which expresses a protein from the paternal allele that promotes cell growth. Analysis of genomic DNA from the patients and their parents by Pinto revealed the selective loss of the maternal chromosome 11 and duplication of paternal chromosome 11 in the pediatric adrenocortical tumors, leading to the continuous high expression of the IGF2 protein and abnormal cell growth.
"With the chromosome 11 abnormality plus the TP53 mutation, you've lost the brakes and stepped on the accelerator at the same time," Zambetti said.
The genomic analysis also yielded the timing of these molecular events. Bioinformaticists Chen and Zhang determined that the chromosomal 17 and 11 abnormalities occur early in tumor development, indicating a fundamental role for these genetic alterations in triggering tumor development.
According to Ribeiro, data on the cancers' genetic landscape offer a highly promising research pathway to understanding the biology and evolution of childhood adrenocortical tumors. "Our focus now will be to determine whether the genomic abnormalities we have distinguished have clinical value in determining the prognosis for these tumors," he said.
In particular, the research team wants to confirm in a larger group of patients that a specific combination of mutations in genes called ATRX and TP53 do lead to more aggressive tumors with poorer prognosis.
The researchers said their studies may also lead to insights into other childhood cancers that also show deregulation of chromosome 11 and over-activity of IGF2, such as rhabdomyosarcoma, Wilms tumor and hepatoblastoma.
The findings also offer considerable promise for improving the treatment of childhood adrenocortical tumors. The study reveals tumor cases with more chaotic molecular changes that will require a different treatment approach. "A key to improving treatment will be using the new genomic knowledge to develop mouse models that would enable more systematic testing, not only of existing therapies, but new ones," Zambetti said.
Source: St. Jude Children's Research Hospital


![WGS for rare disease diagnosis [eBook]](https://d3bkbkx82g74b8.cloudfront.net/eyJidWNrZXQiOiJsYWJyb290cy1pbWFnZXMiLCJrZXkiOiJjb250ZW50X2FydGljbGVfcHJvZmlsZV9pbWFnZV84MmRlM2UyYjA5M2Q3ZTYwOTI3Zjc1YTRjOWU2N2RmMjkzMThjMTJkXzI1MDcucG5nIiwiZWRpdHMiOnsidG9Gb3JtYXQiOiJqcGciLCJyZXNpemUiOnsid2lkdGgiOjcwMCwiaGVpZ2h0IjozNTAsImZpdCI6ImNvdmVyIiwicG9zaXRpb24iOiJjZW50ZXIiLCJiYWNrZ3JvdW5kIjoiI2ZmZiJ9LCJmbGF0dGVuIjp7ImJhY2tncm91bmQiOiIjZmZmIn19fQ==)