The most common form of spinal muscular atrophy (SMA), a rare genetic disease, is caused by mutations of chromosome 5 in the gene, SMN1. SMN1 encodes for the protein, SMN, which translates to "survival of motor neuron." SMN appears to be responsible for the normal function of motor neurons in the spine that control voluntary muscle activity. Nearby SMN2 genes, depending on the number present, can partially compensate for the lack of SMN1 genes. There is also a rare form that is non-chromosome 5-linked SMA.
The majority of the symptoms these patients experience are related to muscle weakness in muscles near the center of the body (shoulders, thighs, upper back, hip). When the muscles involved in breathing and swallowing are affected, the prognosis is poor, resulting in death before two years of age. Due to the high variability in the age when symptoms begin, chromosome 5 SMA is broken down into four types ranging from mild to severe. Type 1 is seen at birth or in infancy and has the poorest prognosis. Types 2 and 3 develop during childhood and type 4 in teens and adults. Patients who develop SMA later in their teens or adulthood have milder and better motor functioning. The age of onset depends on the amount of functioning SMN the patient has, which correlates with the amount of SMN2 genes; the more functioning SMN, the later the onset.
There are currently no cures for SMA; however, on May 24, 2019, the FDA approved a novel gene therapy drug, Zolgensma, to treat SMA in children under two years of age. It is an adenovector-based gene therapy that targets the SMN1 gene and replaces it with a fully functioning copy into the affected motor neurons.
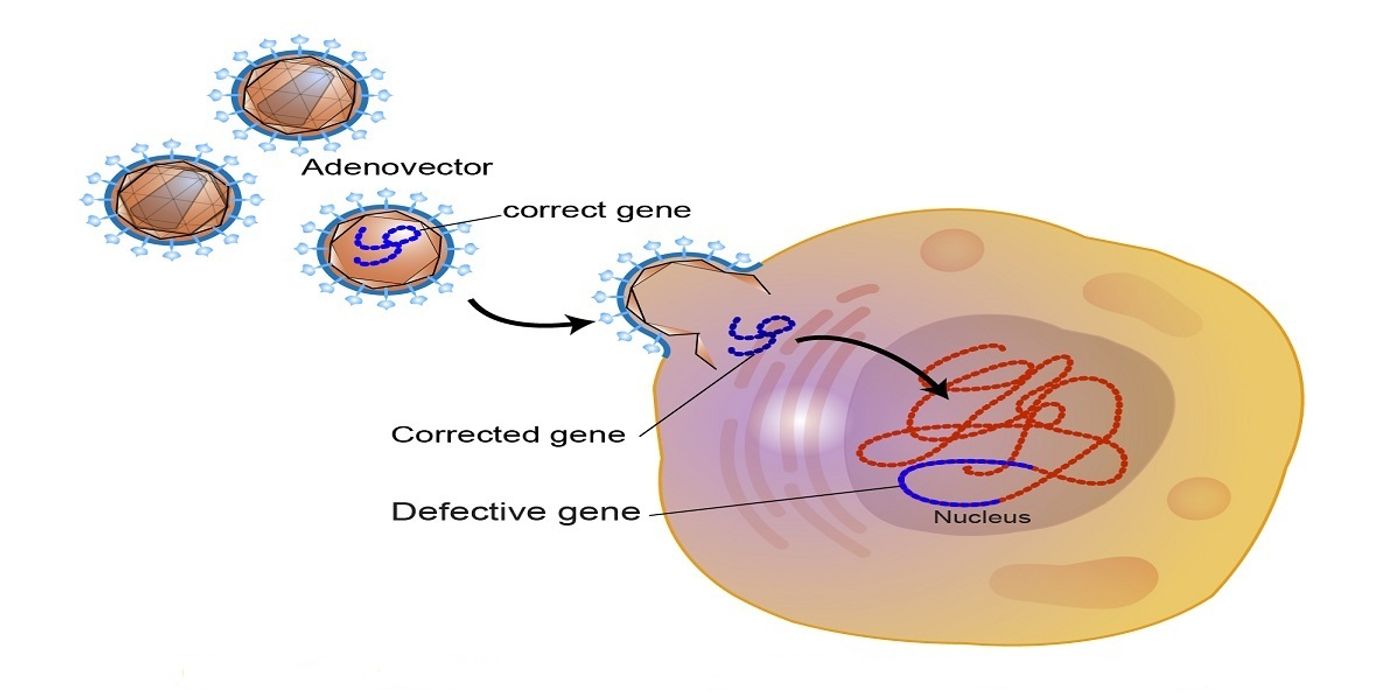
Figure 1. Example of Adenovector-Related Gene Therapy. Credit: National Human Genome Research Institute
Infants receiving this treatment require a one-time IV dose based on their weight. Its effectiveness has been evaluated in one completed clinical trial and one that is ongoing. Significant improvement in meeting milestones such as the ability to sit up without support has been reported in infants with SMA when compared to those not receiving the treatment. The most common side effect is liver damage, which has to be monitored for at least three months after the procedure.
The FDA pushed Zolgensma through as a fast track, breakthrough therapy designated as priority review and an orphan drug. It also awarded the manufacturer, AveXis, Inc., with a rare pediatric disease voucher. There are few treatments available for some rare pediatric diseases, and these programs have been set up by the FDA to encourage research to find better treatments or cures for those diseases that currently have few options.