Developing a new, specialized treatment for autoimmune disease that doesn’t shut down the entire immune system has stumped scientists for years. Designing such a treatment could prevent disorders like rheumatoid arthritis and type one diabetes without leaving the body open to infection from invading pathogens. So what makes an immune cell “snap” and turn against the body’s own “self” cells?
Researchers from the Okinawa Institute of Science and Technology (OIST) Graduate University began their study with this goal in mind: identify the molecular difference between a healthy immune cell and an immune cell involved in autoimmune disease development. They started with a specific type of immune cell, called T helper 17 (Th17) cells. Th17 cells, study author Professor Ishikawa says, influence the body in three main ways: “first to maintain a healthy gut and second to deal with bacterial and fungi infections. The third is their toxicity leading to autoimmune diseases, which is something we want to avoid."
Ishikawa and his team, ideally, would find a way to prevent the activity of self-harm-inflicting Th17 cells without muting the activity of “good” Th17 cells, those fighting infection and keeping the gastrointestinal tract functioning and healthy. A transcription factor called JunB was ultimately discovered to be involved with the immune cell transition from normal to self-inflicting, making JunB the primary target.
The researchers analyzed and searched through nearly three hundred different transcription factors until they identified JunB as the protein they were looking for. They found that JunB prevents certain receptors from being displayed on the surface on Th17 cells, preventing the activation of “bad” Th17 cells.
The receptors thought to be involved in a Th17 cell being driven to autoimmune tendencies are immune messengers, a chemical called interleukin (IL)-23. This immune messenger is simultaneously capable of activating Th17 cells to fight infections and inducing the cells to contribute to an autoimmune reaction. IL-23 activity begins when the chemical binds to its receptor on the surface of Th17 cells, so the seemingly sensible solution would be to prevent IL-23 receptors from being displayed.
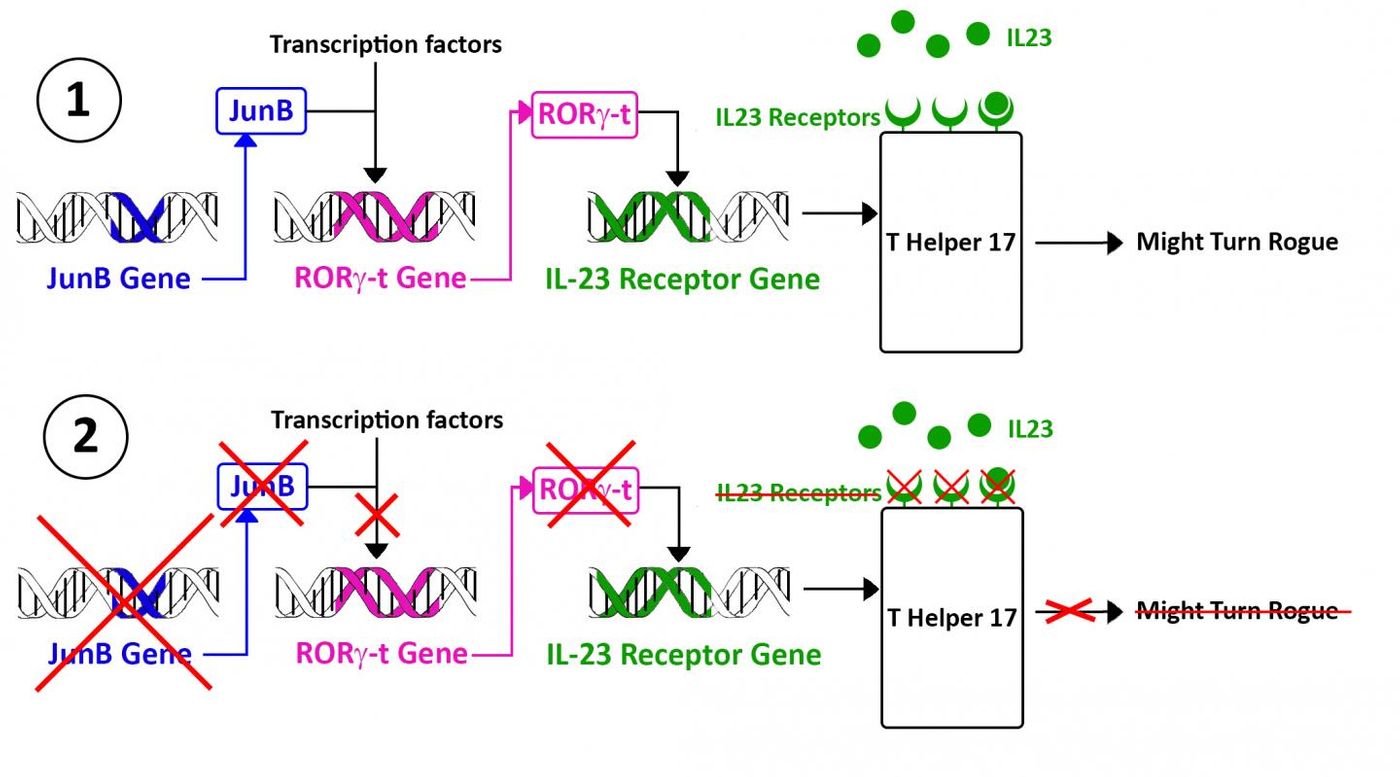
The relationship between JunB, IL-23, and Th17 cells, the researchers concluded, must be unique to autoimmune pathology, because Th17 cells with no JunB still participated normally in healthy immune activity, as seen in mouse models lacking the JunB gene that were “incapable of developing Th17 cell-related autoimmune diseases.” After more study, they saw that JunB influences IL-23 receptor display through a third-party transcription factor, Rory-t.
"Previously, several transcription factors such as RORγ-t have been found essential for all T Helper 17 cells." Ishikawa said. "If we use these molecules as targets for therapy, all T Helper 17 cells will be affected, toxic or not. However, JunB seems to be critical only for toxic T Helper 17 cells: this would allow us to develop a pinpointing, more selective therapy."
Source: Okinawa Institute of Science and Technology (OIST) Graduate University