Recent years have seen an incredible rise in the research and discussion of neuroinflammation, especially in regards to Alzheimer’s disease (AD). However, even though non-steroidal anti-inflammatory drugs (NSAIDs) have no effect on AD progression and it is not possible to treat AD by eating foods with anti-inflammatory properties, that doesn’t mean a more targeted anti-inflammatory treatment would not be beneficial, and that is exactly what Olmos-Alonso et al. are exploring in their recent paper published in
Brain.
When we talk about neuroinflammation, what we are really saying is that there is proliferation and activation of microglia, a specific cell type in the brain that acts as its immune system. Just like in the rest of the body, an acute inflammatory response in the brain is good because it means your microglia are attacking whatever invader they have encountered. Inflammation, in the brain or any other part of the body, becomes a problem when it is chronic. Microglia, as the brain’s immune system, are drawn to the amyloid plaques that are characteristic of AD to try and clear them out. Unfortunately, the plaques are so dense that the microglia cannot clear them, which leads to a chronic inflammatory state as the microglia are maintained in a state of activation, continually releasing inflammatory signals.
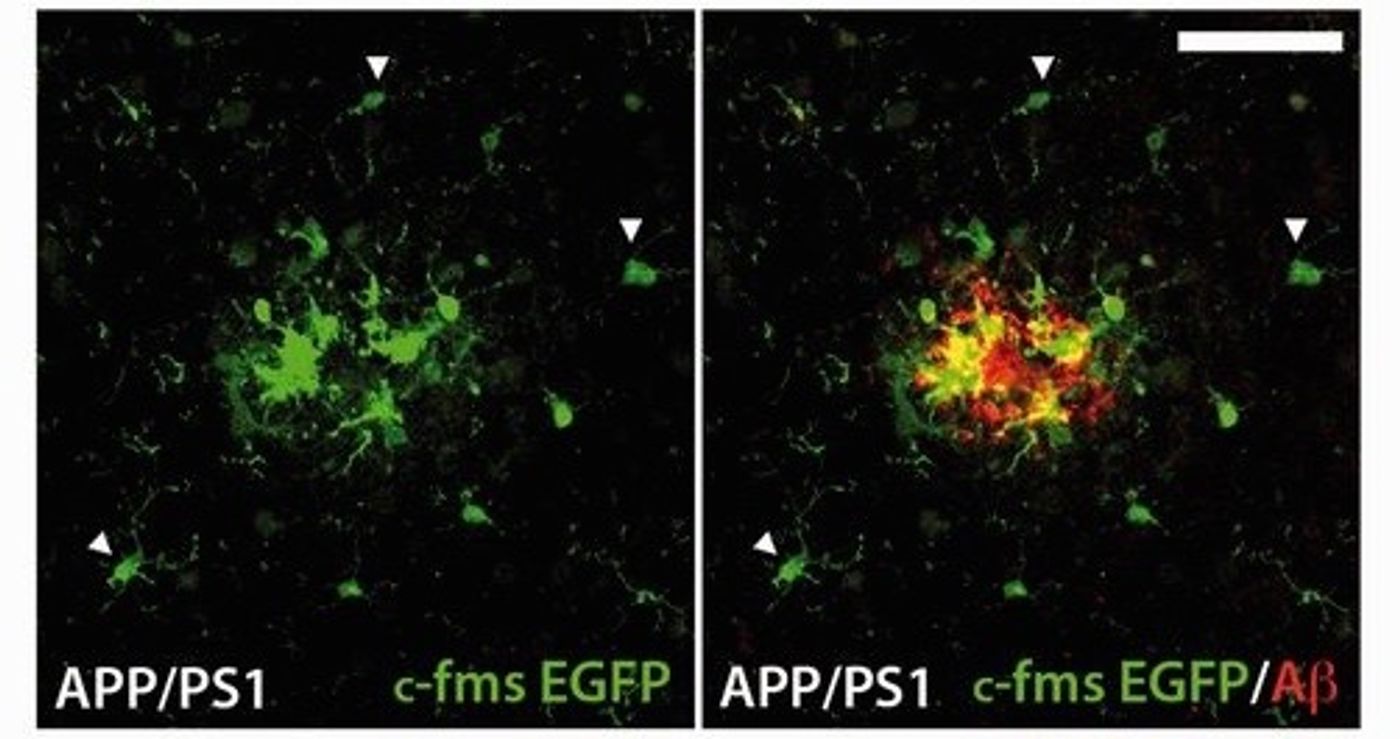
Olmos-Alonso et al. have targeted a key factor in the proliferation of microglia surrounding amyloid plaques, colony-stimulating factor 1 receptor (CSF1R). Elevated expression of CSF1R is positively correlated with microglial activity near amyloid plaques and disease severity. This microglia-specific receptor is at the top of a proliferation signal cascade, and gene expression analysis has shown that key downstream components in this pathway are also upregulated in postmortem AD brain tissue compared to healthy controls. These results were also confirmed in a transgenic mouse model of AD. In the transgenic mice, the number of microglia surrounding amyloid plaques increased as the mice aged and the disease progressed. And just like in the human tissue, CSF1R and the other components of its proliferation pathway were upregulated in the transgenic mice as compared to the wild-type controls.
What does this mean? Why is this important? This paper is interesting and relevant because CSF1R is a good, specific, druggable receptor, ideal for the development of a small molecule therapeutic. As a starting point, the researchers used GW2580, a tyrosine kinase inhibitor that inhibits CSF1R activity. When orally given to the transgenic AD mice, it prevented microglial proliferation, slowed disease progression, and improved behavior and cognition. Importantly, GW2580 treatment prevented synapse loss. Synapses are how neurons communicate to each other, and when a neuron loses its means of communication, it dies. Even though GW2580 has no effect on amyloid plaques, the recovery of synapses can be considered a disease-modifying effect. Having a disease-modifying AD drug is the goal. We don’t want to just cover up symptoms, like a cholinesterase inhibitor does, we want to modify the progression of the disease. The strength in this paper comes from the fact that they attacked the issue of inflammation in a targeted and precise way. It will be exciting to see if this line of research makes it to the clinic.