Abstract
One of the primary objectives for the application of flow cytometry in any testing environment should be measurement assurance, i.e., the generation of reliable and reproducible results. This goal can only be realized using multiple controls during each stage of the measurement process: instrument qualification, instrument monitoring, panel design, testing, and assay monitoring. There have been numerous global initiatives focusing on measurement consistency in flow cytometry. This review will provide a high-level overview of the major global initiatives related to achieving measurement assurance in flow cytometry and the current recommendations for best practices. The primary focus of this manuscript will be on the controls, calibrators, and standards used in flow cytometry.
Introduction
The flow cytometry community is witnessing significant advances in instrumentation, reagents (fluorescent probes), and software analysis tools such as artificial intelligence (AI) for single-cell analysis (1). The use of flow cytometry is expanding beyond biomedical research and clinical settings into drug discovery and development, cell and gene therapy, transfusion medicine, hematopathology, botany, marine and environmental biology, food sciences, and animal husbandry. The importance of generating accurate flow cytometry data is only increasing, especially as it becomes indispensable to applications such as measurable residual disease (MRD) in oncology, evaluation of biomarkers supporting drug development, and cell therapy manufacturing. In this way, flow cytometric data enhances Investigational New Drug (IND) applications and contributes to key decisions regarding therapeutic regimens. The critical quality attributes (CQA) in cell therapy manufacturing include flow cytometric evaluation. As the new applications for flow cytometry expand, the requirement for high-quality data also increases. This, in turn, creates the need to redefine best practices, educate users regarding best practices, and finally, ensure these are implemented. Increased rigor in measurement is widely recognized as critical to advancing biosciences and biotechnology.
The interest in high-quality and combinable data sets is not new to the field of flow cytometry (2, 3). As the technology has evolved and as the applications have expanded, various consortia and working groups have recognized the need to establish new best practice guidelines and refine existing guidance documentation. Their aims have been to identify the underlying obstacles in the field and put forth recommendations to overcome existing limitations in generating standardized and uniform results. The AIDS Clinical Trial Group (ACTG) was one of the first groups to conduct an inter-laboratory study and put forth best practices recommendations in the 1980s with the goal of obtaining consistent data for all testing laboratories (4).
In 1996, the International Society of Hematotherapy and Graft Engineering (ISHAGE) established guidelines for CD34+ progenitor enumeration, a procedure that is now used to evaluate hematopoietic stem cell mobilization, among other clinical applications (5). The EuroFlow Consortium began to publish their recommendations for leukemia and lymphoma testing in 2007 (6). Recommendations specific to the validation of flow cytometric methods were first published by the American Association for Pharmaceutical Scientists (AAPS) first in 2011 and again in 2013 by a collaborative effort between the International Clinical Cytometry Society (ICCS) and the International Society for Standards in Hematology (ISLH)(7, 8). In 2021, the Clinical and Laboratory Standards Institute’s (CLSI) Guidance H62 for the validation of flow cytometric methods was published (9). More recently, the Extracellular Vesicle (EV) Flow Cytometry Working Group (EV FC WG) published recommendations around the measurement of EV in 2022. (10). These recommendations and guidelines cover most aspects of measurement, from pre-analytical steps (including sample collection, storage, and processing), to instrumentation (including initial qualification, calibration, standardization, and daily monitoring) and assay design (including panel design, sample staining, essential controls, gating, and reportable results) (Figure 1).
Measurement Assurance in Flow Cytometry
Measurement assurance is a structured process designed to ensure that measurements are adequate for their intended use. It is accomplished by treating the assay as a measurement process. First, different elements that contribute to the measurement are defined, and then processes for each element are designed (Figure 1). Next, quality assurance (QA) and quality control (QC) practices are established to maintain a controlled environment that can produce high-quality, robust, and validated measurements for cell characterization, and which is able to consistently generate reliable data over a long period of time. Finally, key performance indicators for each element are defined along with processes for maintaining and monitoring performance.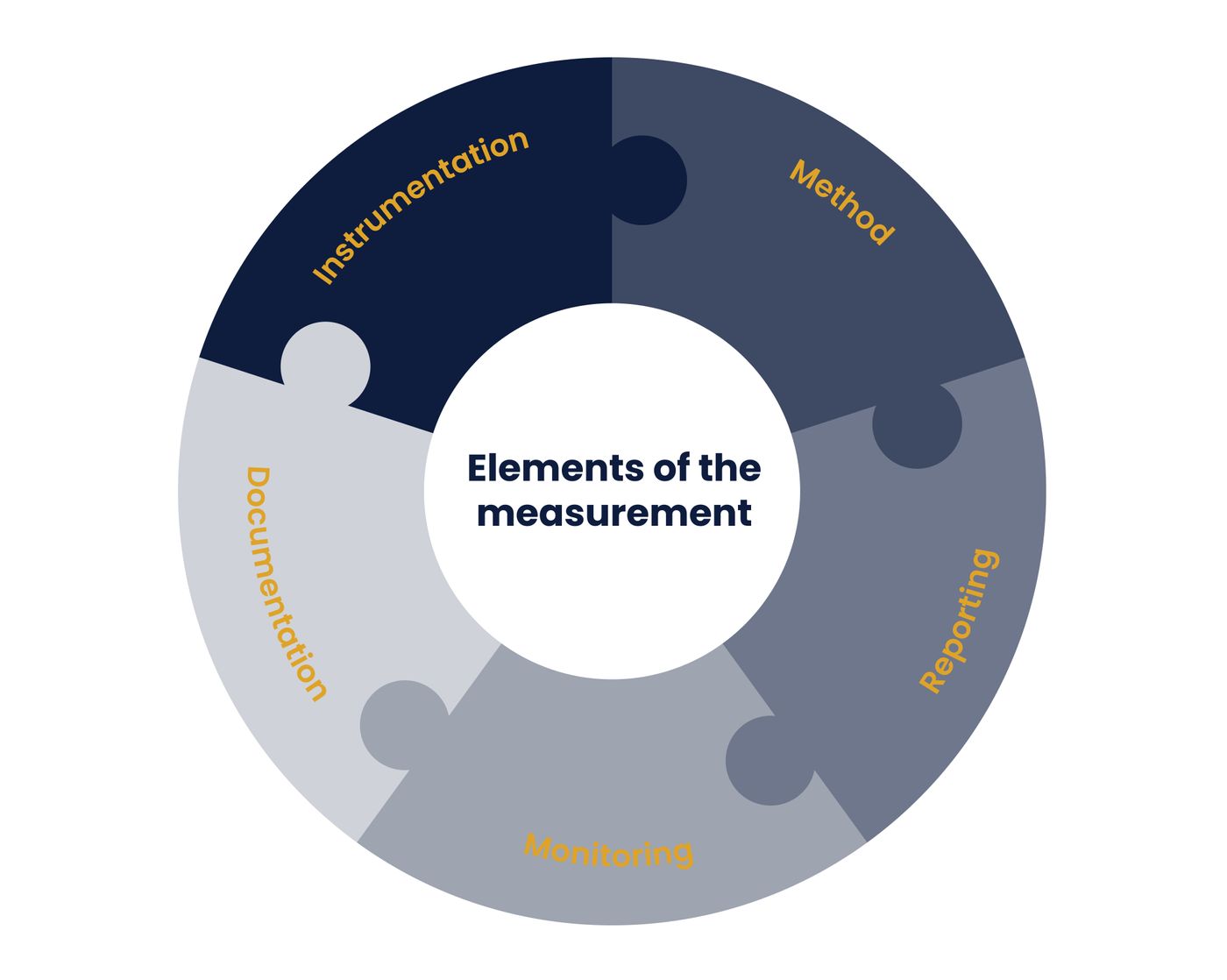
Figure 1. Measurement Assurance in Flow Cytometry. All elements which are part of the process of generating the measurement contribute to assuring that the measurement will be adequate for its intended use.
In order to achieve comprehensive measurement assurance in flow cytometry, QA and QC procedures span from the initial instrument qualification to assay development and validation. These activities are considered the pre-analytical or pre-examination activities. During testing or the examination phase, separate QA and QC procedures should be initiated in order to monitor the environment, the instrument, the reagents, and the assay, as well as staff training. The purpose of these QA and QC procedures is to ensure that the instrument continues to perform in the qualified state, new lots of reagents are comparable to existing lots, the assay continues to perform in the validated state, and that current and new staff continue to be fully competent.
Measurement assurance activities in flow cytometry require the use of both calibrators and controls (Tables 1 and 2). The calibrators are generally beads with known values for chosen characteristics and are used for instrument evaluation, adjustment, and monitoring. Beads may be hard-dyed and fluoresce over a broad spectral range, or they may be surface- labeled and spectrally matched to the fluorochromes used to stain cells. Spectrally matched beads are used for standardization and calibration across instruments that have different lasers and/or filters. Controls are generally understood to be biological samples intended to monitor all aspects of the assay and sample processing. When processed in the same manner as the intended use samples, the controls should produce results within a pre-defined range based on the performance characterization of the method. When deciding the best controls for a given process (i.e., cells or beads, spectrally matched or broad-spectrum), the objectives of each process and the objectives of the control procedure must be fully understood.
Instrument
At each stage of an instrument’s lifecycle, multiple processes are executed to ensure that results from the instrument will have the quality required for their intended use. Instrument qualification, calibration, standardization, and subsequent routine monitoring each provide different degrees of certainty related to instrument performance and, in turn, measurement assurance.
Table 1. Instrument Calibrators. A variety of particles are used during the various phases of instrument QA and QC
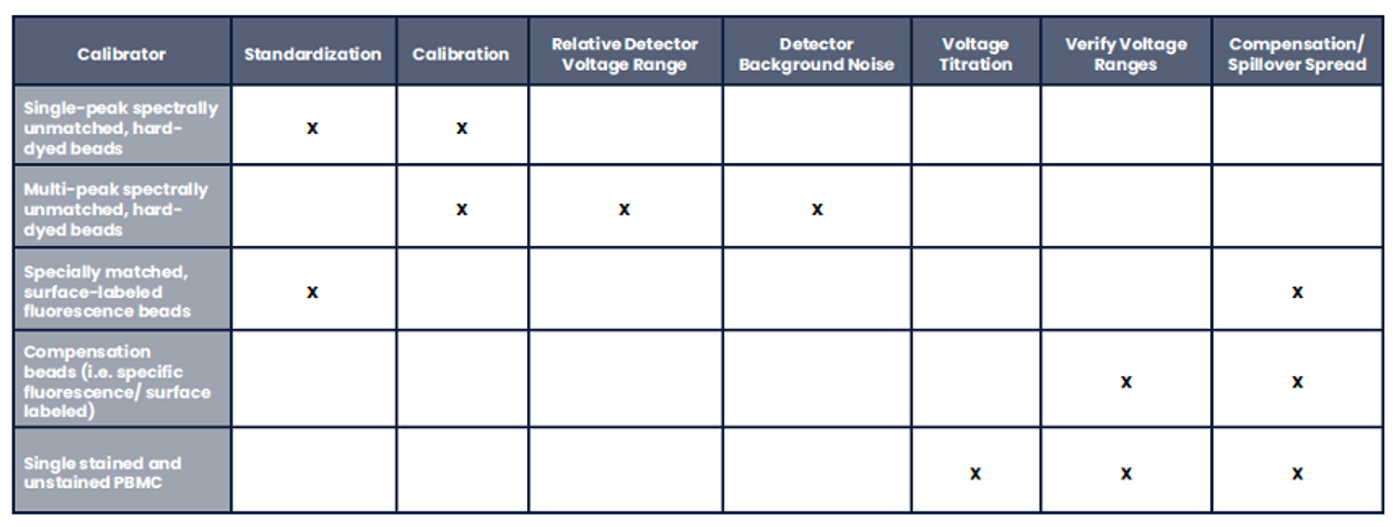
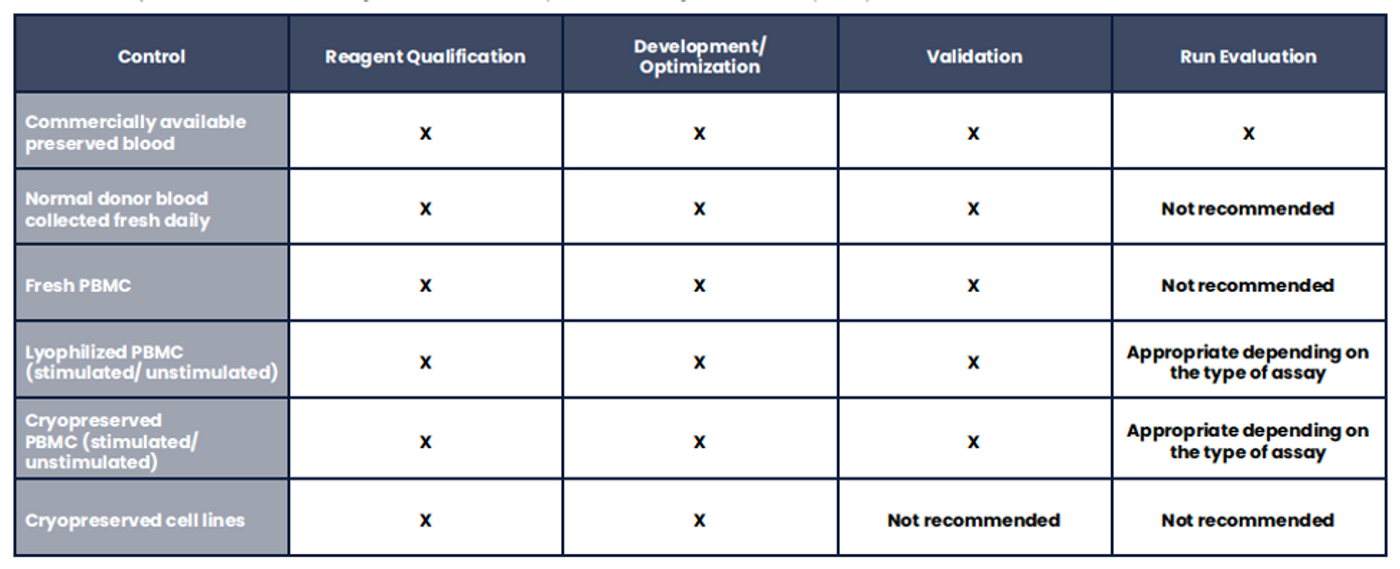
Table 2. Assay Control Materials. Biological controls are required at all stages of the assay lifecycle.
Qualification
When first placing a new instrument in the laboratory, the testing laboratory should qualify the instrument. The level of rigor for the instrument qualification procedure and the associated documentation will depend on the regulatory environment of the testing laboratory. Instrument qualification is a multistep process that aims to ensure that reliable data can be generated from each assay throughout instrument’s lifecycle.
Best practices for instrument qualification are described in depth in CLSI H62 and by Green et al. (9, 11). Briefly, instrument qualification involves three phases: 1) Installation Qualification (IQ); 2) Operational Qualification (OQ); 3) Performance Qualification (PQ). IQ entails assessing the facilities’ requirements for the instrument, for example, hardware, software, physical space, and electrical requirements. During OQ, the basic functionality of the instrument is verified, including but not limited to the software functionality, system alerts, optical precision and sample acquisition. IQ and OQ are often performed by the manufacturer during installation to demonstrate that the instrument and its associated software are installed and functioning per the manufacturer’s specification and the user’s requirements. PQ is a more extensive process, which is most often performed by the testing laboratory. PQ includes instrument optimization and extended longitudinal performance (Table 3).
Optimization
Optimization examines a wide array of critical instrument parameters: the efficiency and performance of optical filters, dichroic mirror reflection and transmission, the timing of lasers (laser delay), laser power, the length of time set to collect instrument signals (e.g., window extension), amplifier linearity, electronic noise, and synchronization of the area and peak height signals (e.g., area scaling factor) (12, 13).
Table 3. Performance Qualification. A variety of instrument functionality is evaluated during Performance Qualification.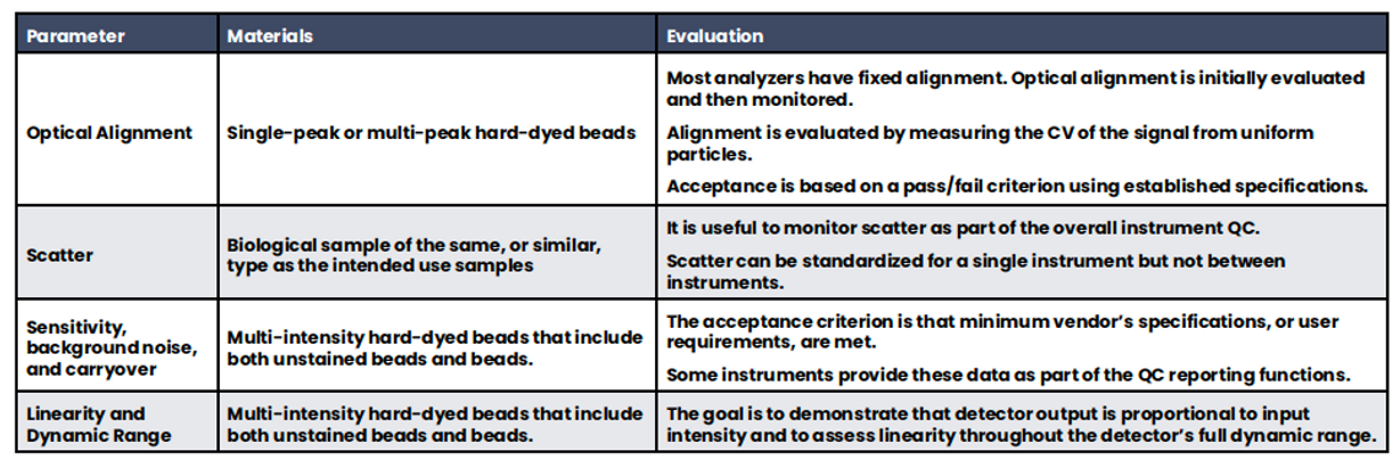
Calibration
Detailed procedures for instrument calibration are described in depth by Perfetto et al (13). Briefly, calibration is accomplished by measuring fluorescence over a range of photomultiplier tube (PMT) voltages in order to determine the PMT voltage range and linearity. These procedures also apply to other excitation and detection modalities, including solid-state sources and spectral analyzers. Instrument calibration is required for the objective and quantitative comparison of inter-inter instrument and inter-laboratory longitudinal data.
Daily Monitoring
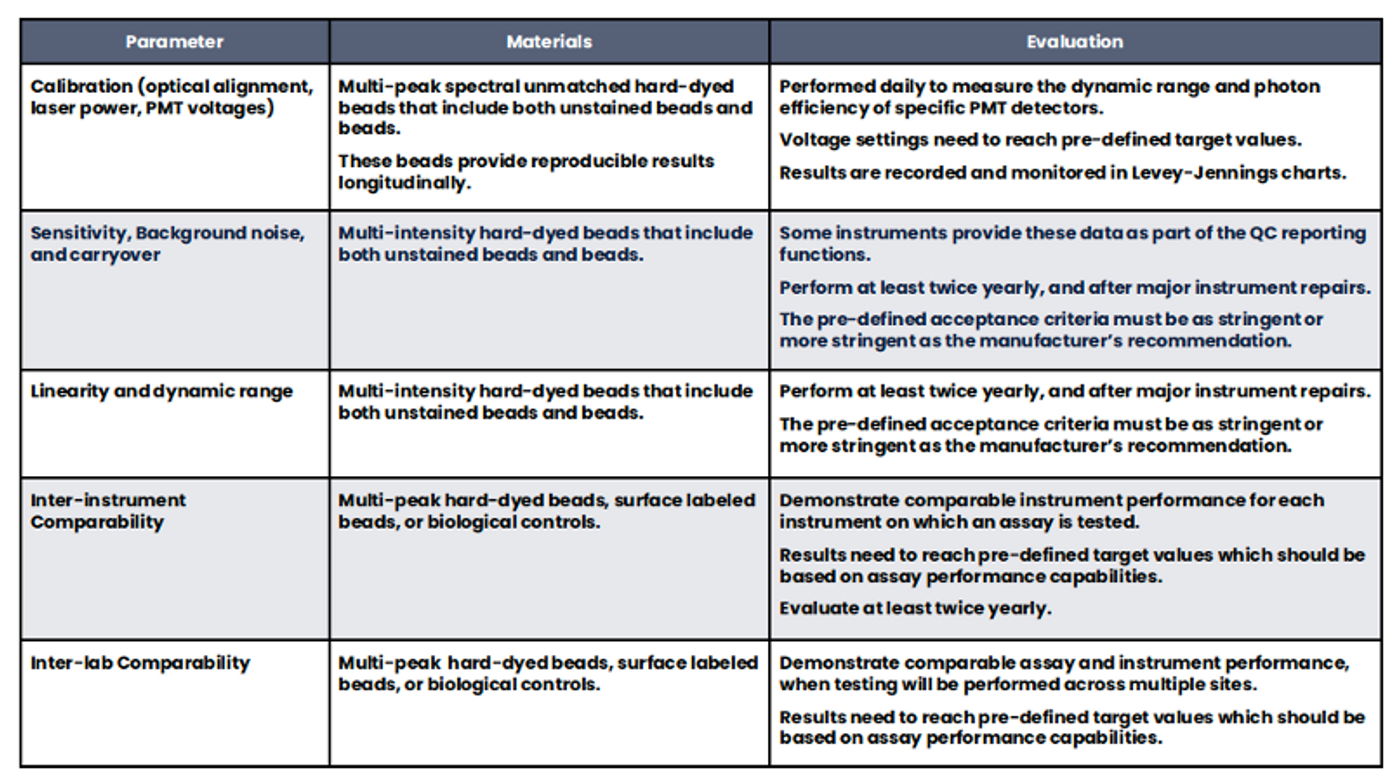
Once the instrument has been qualified and put into use, it should subsequently be operated under QA/ QC conditions to maintain the instrument within predetermined bounds and ensure that results will vary only within certain pre-defined limits (Table 4). The daily QA/QC practices, as described in CLSI H62, are intended to monitor instrument variation to verify and document that the system is suitable for daily use (9). In addition, data from the daily QA/QC can be used to track trends in instrument performance, such as detector sensitivity or changes in the filters, thereby providing information about potential future issues or the need for repairs.
Assay
Development, Optimization, and Validation
The development of a multi-color panel is an iterative process. Best practices for multicolor panel design (assay development) and validation are well described in the literature (9, 14, 15). Typically, both commercially available biological controls such as preserved whole blood products or lyophilized PBMC and intended use samples are used. During panel design, biological controls can be used to screen monoclonal antibodies (mAb) and demonstrate the overall assay specificity (i.e., that the assay measures what it was intended to measure).
Another important step in panel design is to optimize the fluorescence compensation and minimize spillover, as described in (16). Compensation beads or single stained cells can be used for this purpose. Because of the differences in light scatter and autofluorescence between cells and beads, there is considerable debate whether cells or beads are preferable for creating the compensation matrix and spillover spread matrix (SSM).
During analytical method validation, biological controls are useful when characterizing inter-assay and inter- laboratory performance, particularly when sample stability is limited (7, 9). Nonetheless, commercially available biological controls have several drawbacks. Their primary limitation is that they are not provided with meaningful target values beyond the primary lymphocyte subsets. Thus, they cannot be used to assess true accuracy and linearity, as discussed in depth in CLIS H62. Despite this limitation, the commercially available biological controls are well suited for inter-assay and inter-laboratory comparisons (9). Other limitations are discussed below.
Daily Testing and Routine QC
Biological process controls should be included in each analytical run, especially when flow cytometry is used to support patient care and treatment or as biomarkers in longitudinal clinical trials. When used in this capacity, the performance of the process controls will be one of the primary criteria for evaluating and accepting each analytical run. If the process controls are within the pre- established ranges, there is assurance that the assay was performed according to the SOP, the reagents have not degraded, and the instrument is operating within predefined specifications. Biological controls are also a critical component of other QC processes in the flow cytometry laboratory; for example, they are often used to evaluate new lots of mAb, for staff training, and as a part of troubleshooting activities.
The ideal process control material would be a biological control with similar properties to the intended use samples, delivered equal or better assay precision compared to the intended use samples, and had a reasonably long shelf-life. Various process control materials have been used in different laboratories, such as; commercially available preserved blood, commercially available lyophilized or cryopreserved PBMC, in-house generated cryopreserved PBMC, in- house generated cell line banks, and fresh normal donor samples (Table 4). Each of these materials presents advantages and disadvantages, and unfortunately a perfect biological control material does not exist to date. The general consensus is that the field could benefit greatly from the introduction of novel, biologically- relevant process controls.
Commercially available preserved blood controls are suitable biological process controls for basic methods such as assays which monitor only the major lymphocyte populations (Total T cells, CD4 T cells, CD8 T cells, B cells, NK cells). The major limitation of these products is limited stability, resulting in the laboratory’s need to frequently re-establish new ranges for each new lot of biological control material. This is costly as it is a time-consuming process that uses large quantities of reagent and ties up instrument time. Unfortunately, for these reasons, some laboratories choose to reduce their usage of this valuable quality monitoring tool in order to reduce operating costs. The preserved blood products have additional limitations for higher-dimensional assays that evaluate more extensive cell subsets. Their primary drawback is that not all of the cell populations evaluated in higher dimensional assays will be detectable in the control material. As a result, the entire assay is not controlled for using this type of control. An additional disadvantage is that the commercial processing of these biological controls can result in a loss of more labile antigens, which can result in decreased resolution of cell subsets expressing low levels of these antigens. In these situations, the control material will may greater variability than the intended use sample, which will decrease confidence in the reported data and could also lead to the unnecessary cancellation of an analytical run.
Lyophilized PBMC controls have greater stability than preserved blood controls; however, they are not ideal in assays designed for whole blood or bone marrow matrices as they are not similar to the intended use samples and would not control for cell lysis steps.
Table 4. Extended Performance Qualification. Performance Qualification does not end after successful installation, it includes routine QA/QC and extends throughout the lifecycle of the instrument. The purpose is to ensure that the qualified instrument continues to perform as expected.
There are few commercially available biological controls for leukemia and lymphoma immunophenotyping panels. Many laboratories will use internal positive and negative populations for reagent monitoring. Although this is an accepted practice, it does not allow the laboratory to monitor the assay performance over time, which is an extremely valuable QA/QC tool for adequately detecting reagent degradation or instrument decline.
Some laboratories will create their own QC material with cell lines or ex vivo PBMC activation. Maintaining cell lines is a labor-intensive practice. If the laboratory does not have adequate quality practices for consistent cell line propagation, viability monitoring, cryopreservation, and thawing, the in-house generated, cell line-derived process controls run the risk of introducing variability to the quality monitoring process.
The use of healthy donor samples as process controls is another common practice; however, due to intra- and/ or inter-donor variability, healthy donor samples are not ideal process controls. A comparison of preserved commercial controls to healthy donor samples reveals that assay variability appears higher with healthy donor controls than commercially available preserved blood controls (Figure 2). This artificial increase in assay variability comes not from the assay but from the variability of the control material due to the intra- and/ or inter-donor variability of healthy donors.
 Figure 2. Assay Monitoring. The performance of an IVD assay for peripheral blood T cells was monitored with a commercially available whole blood control (orange dot) and healthy donor whole blood (navy square). Both QC materials were included in the same analytical runs. Assay variability appears higher with the healthy donor whole blood control.
Figure 2. Assay Monitoring. The performance of an IVD assay for peripheral blood T cells was monitored with a commercially available whole blood control (orange dot) and healthy donor whole blood (navy square). Both QC materials were included in the same analytical runs. Assay variability appears higher with the healthy donor whole blood control.
In summary, the ideal process control would be stable, yield consistent results, and, most importantly, not introduce its own variability. It is important that the variability observed during the monitoring process comes from the system being monitored, for example, the instrument, the reagent, the assay, the analyst, and the laboratory, not from the control material itself.
Inter-laboratory Studies and Assay Transfers
There are numerous different scenarios where flow cytometric methods are conducted in more than one testing laboratory (17). For example, in translational medicine, inter-laboratory studies are becoming more and more common. In drug development, assays are frequently transferred from one laboratory to another, either within the same institution or between institutions. Flow Cytometry used in global clinical trials requires that testing be conducted at multiple locations owing to both limited specimen stability and high shipping costs. In cell and gene therapy studies, the flow cytometry methods used to establish CQA for product release are also transferred from one laboratory to another as the therapy progresses along the drug development pathway.
To generate consistent data sets from multiple instruments and multiple laboratories, the instruments and the assays should be standardized. In all laboratory settings, the instrument and the assay should be routinely monitored using control material per the
established QA/QC practices, however, in inter- laboratory studies and assay transfers, these QA/QC processes are mandatory in order to ensure the delivery of high-quality, consistent, and combinable data.
Moreover, many inter-laboratory studies, particularly longitudinal, global clinical trials, include the participation of laboratories located in different countries that are often geographically distant. These circumstances put additional requirements on the control material, such as the capacity to be shipped at ambient temperatures. The biohazard classification of the control material is an additional consideration given that some countries have import licensing procedures for biohazardous material, which can be cumbersome and difficult to navigate.
Discussion
In the past several years, the flow cytometry community has witnessed revolutionary advances in instrumentation, novel fluorophores, and data analysis approaches, such as those that now include AI (1). Some of the greatest excitement in flow cytometry is currently focused on the potential of implementing automated data analysis tools and unsupervised gating approaches. Many cytometrists think that the automation and standardization of data analysis is an essential requirement for ushering in the new era of high-dimensional, quantitative cytometry. However, one critical point that is often missing from discussions surrounding the future of data analysis is the need to demonstrate that the data submitted to the analysis tools are of suitable quality. The old cliché of garbage-in, garbage-out comes to mind. Thus, the implementation of the QA/QC processes for the instrument and the assay discussed herein should be broadly implemented, even in non-regulated testing laboratories.
Process controls are crucial in the implementation of QA/QC programs. In flow cytometry, the best process controls are biological controls. They are required during assay development, optimization, validation, and monitoring as well as in inter- instrument and inter-laboratory standardization. Unfortunately—and despite the well-documented need for improved biological process controls— the development of biological control material for flow cytometry has not progressed in parallel with the advances in instrumentation, reagents, and software (7, 8, 18, 19). The CLSI guideline H42-A2 (19) takes a conservative approach, stating, “There are at present no standards which can be used to check the accuracy of flow cytometric test results.” At CYTO 2018, a workshop on the topic of control cells was held with the intention of defining the pathway to overcome the challenge issued by the lack of readily available biological control materials (18). One conclusion from this workshop was that biological control material for flow cytometry needs to evolve in tandem with the instruments, reagents, and software approaches. Control cells are important yet remain challenging for the field of flow cytometry. There remains an unmet need for innovative new solutions for this critical aspect of generating high- quality results.
Bibliography
- Mitra-Kaushik S, Mehta-Damani A, Stewart JJ, Green C, Litwin V, Gonneau C. The evolution of single cell analysis and utility in drug development. The AAPS Journal. 2021, The AAPS Journal (2021) 23:98 DOI: 10.1208/s12248-021-00633-6.
- Gonneau C, Wang L, Mitra-Kaushik S, Trampont PC, Litwin V. Progress towards global standardization for quantitative flow cytometry. Bioanalysis. 2021 Sep;13(21):1591-5.
- Kalina T. Reproducibility of flow cytometry through standardization: opportunities and challenges. Cytometry Part A. 2020 Feb;97(2):137-47
- Paxton H, Kidd P, Landay A, Giorgi J, Flomenberg N, Walker E, Valentine F, Fahey J, Gelman R. Results of the flow cytometry ACTG quality control program: analysis and findings. Clinical immunology and immunopathology. 1989 Jul 1;52(1):68-84.
- Sutherland DR, Anderson L, Keeney M et al. The ISHAGE guidelines for CD34+ cell determination by flow cytometry. J Hematother 1996;5:213-26.
- Szczepański T. Why and how to quantify minimal residual disease in acute lymphoblastic leukemia?. Leukemia. 2007 Apr;21(4):622- 6.
- O’Hara, D., Xu, Y. Lianz, E., Reddy, M., Wu, D., and Litwin, V. Recommendations for the Validation of Flow Cytometric Testing During Drug Development: II Assays. Journal of Immunological Methods, 363:120-134, 2011.
- Wood, B., Jevremovic, D., Béné, MC., Yan, M,. Jacobs, P., Litwin, V. Validation of cell-based fluorescence assays: Practice guidelines from the ICSH and ICCS – Part V – assay performance criteria. Cytometry Part B: Clinical Cytometry. 84:315, 2013.
- Validation of assays performed by flow cytometry. 1st ed. CLSI document H62. Wayne, PA: Clinical Laboratory Standards Institute; 2021.
- Welsh JA, Van Der Pol E, Arkesteijn GJ, Bremer M, Brisson A, Coumans F, Dignat-George F, Duggan E, Ghiran I, Giebel B, Görgens A. MIFlowCyt-EV: A framework for standardized reporting of extracellular vesicle flow cytometry experiments. Journal of extracellular vesicles. 2020 Sep;9(1):1713526.
- Green CL, Brown L, Stewart JJ, Xu Y, Litwin V, Mc Closkey TW. Recommendations for the validation of flow cytometric testing during drug development: I instrumentation. Journal of immunological methods. 2011 Jan 5;363(2):104-19.
- Wang, L. and Hoffman, R.A. 2017. Standardization, calibration, and control in flow cytometry. Curr. Protoc. Cytom. 79:1.3.1-1.3.27. doi: 10.1002
- Perfetto SP, Ambrozak D, Nguyen R, Chattopadhyay P, Roederer M. Quality assurance for polychromatic flow cytometry. Nature protocols. 2006 Aug;1(3):1522-30.
- Ferrer-Font L, Pellefigues C, Mayer JU, Small SJ, Jaimes MC, Price KM. Panel design and optimization for high-dimensional immunophenotyping assays using spectral flow cytometry. Current protocols in cytometry. 2020 Mar;92(1):e70.
- Cossarizza, A., et al. (2019). Guidelines for the use of flow cytometry and cell sorting in immunological studies. European journal of immunology, 49(10), 1457-1973.
- Nguyen R, Perfetto S, Mahnke YD, Chattopadhyay P, Roederer M. Quantifying spillover spreading for comparing instrument performance and aiding in multicolor panel design. Cytometry Part A. 2013 Mar;83(3):306-15.
- Cabanski M, Oldaker T, Stewart JJ, Selliah N, Eck S, Green C, Litwin V, Vitaliti A. Flow cytometric method transfer: Recommendations for best practice. Cytometry Part B: Clinical Cytometry. 2021 Jan;100(1):52-62.
- Czechowska, K, et al. Cyt-Geist: Current and Future Challenges in Cytometry: Reports of the CYTO 2018 Conference Workshops. Cytometry Part A 95A: 598–644, 2019
- Enumeration of Immunologically Defined Cell Populations by Flow Cytometry. 2nd ed. CLSI document H42. Wayne, PA: Clinical Laboratory Standards Institute; 2007.