Muscle diseases that have a genetic basis can cause muscles to waste away, and may lead to a premature death. There aren't many treatment options for these disorders, which include multiple sclerosis and other rare diseases like X-linked myotubular myopathy. Gene therapy could be an option for patients, but it's been difficult to get the therapy into the nucleus of cells where the DNA is. Viruses that don't cause disease but can enter cells called adeno-associated viruses (AAVs) are typically used as delivery systems in gene therapy, but for muscle diseases, high doses are required to get enough of them to all the muscle tissue in the body. As such, lots of the AAV ends up in the liver, causing major side effects and even death in some trial volunteers.
Scientists may have found a way to overcome this challenge by creating a new type of AAV, called MyoAAV, which is ten times better at getting into muscle than other viral vectors. Not much ends up in the liver, and gene therapies can be given to patients at doses that are 100 to 250 times lower than those used with different viral vectors in other trials. The researchers are hopeful that serious side effects can now be avoided. The findings have been reported in Cell.
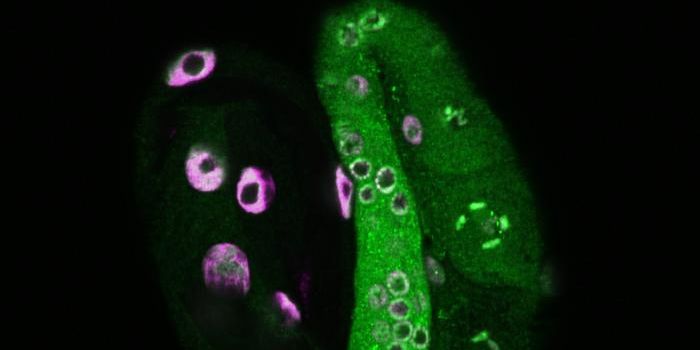
In this comprehensive study, the researchers began by altering AAV's shell or capsid, using an approach they created called Directed Evolution of AAV capsids Leveraging In Vivo Expression of transgene RNA (DELIVER). First, millions of AAV capsids were produced, with a short string of extra amino acids on the receptor AAV uses to bind to cells. Muscle tissue was then exposed to these capsids, and the researchers sequenced the muscle cells to identify the capsids that were best at binding to cells and shuttling the gene-altering therapy inside. Once the MyoAAV viruses were found, the researchers began to test them in animal models.
“Our method is unique because we screened a wide array of capsids and used very stringent selection criteria. We wanted to find capsids that could not only physically enter the cell, but also proceed through different steps of transduction and express their transgenes,” said first study author Sharif Tabebordbar, a Pardis Sabeti lab member at the Broad Institute.
The MyoAAV vector was successfully able to direct therapeutic genes or the components of the CRISPR-Cas9 gene editor to muscle cells in their models. There was improvement in muscle function in mouse models of Duchenne muscular dystrophy,and X-linked myotubular myopathy. In Duchenne's, a gene called dystrophin is dysfunctional. MyoAAV ferrying CRISPR-Cas9 led to repairs in dystrophin in muscle tissue and improvement in strength and function in the mice.
The researchers collaborated with the Alan Beggs lab at Boston Children’s Hospital to investigate how MyoAAV could help treat X-linked myotubular myopathy (XLMTM), which is usually lethal to a mouse model by ten weeks of age. When six of these mice were treated with MyoAAV, they lived as long as normal mice, while mice treated with AAV only survived for nine weeks.
The gene therapy was also able to enter cells in nonhuman primates and human muscle cells in culture.
“We have developed a family of viral capsids that exquisitely targets skeletal muscle, that we’ve been able to test through multiple preclinical stages and in multiple disease models,” said Sabeti. “We went through many, many preclinical steps to find something that may potentially be ready for the clinic in the near future.”
Experienced research scientist and technical expert with authorships on over 30 peer-reviewed publications, traveler to over 70 countries, published photographer and internationally-exhibited painter, volunteer trained in disaster-response, CPR and DV counseling.