News about the completed work of The Cancer Genome Atlas (TCGA) came out earlier this year characterizing 33 adult tumor types into a cancer index based on complex molecular analyses and carefully made conclusions to inform clinical diagnosis and patient care. While pan-cancer analyses have been done for adult cancers, a parallel of that, with broad scope, had not been done for pediatric cancers. Pediatric cancers are harder to study because of the small number of patients. Despite this, it has been reported that approximately 20% of young cancer patients still die from their disease; improving survival rates and studying these cancers continues to be imperative. Additional layers for surviving children include suffering from severe long term side effects of treatment (surgery, chemo- or radio- therapies) and the causation of therapy induced secondary cancers.
You might ask why data from adult cancer research can’t be extrapolated and applied to young patients, like altering pharmaceutical concentration based on weight or age. In some instances, evaluating and finding trends for tumor markers and genetic changes between adult and pediatric cancers can yield relevant prognostic or diagnostic information; however, many of the differences lie in the developing cancerous tissue type where adult cancers are primarily of epithelial tissues and pediatric cancers occur in mesodermic tissues.
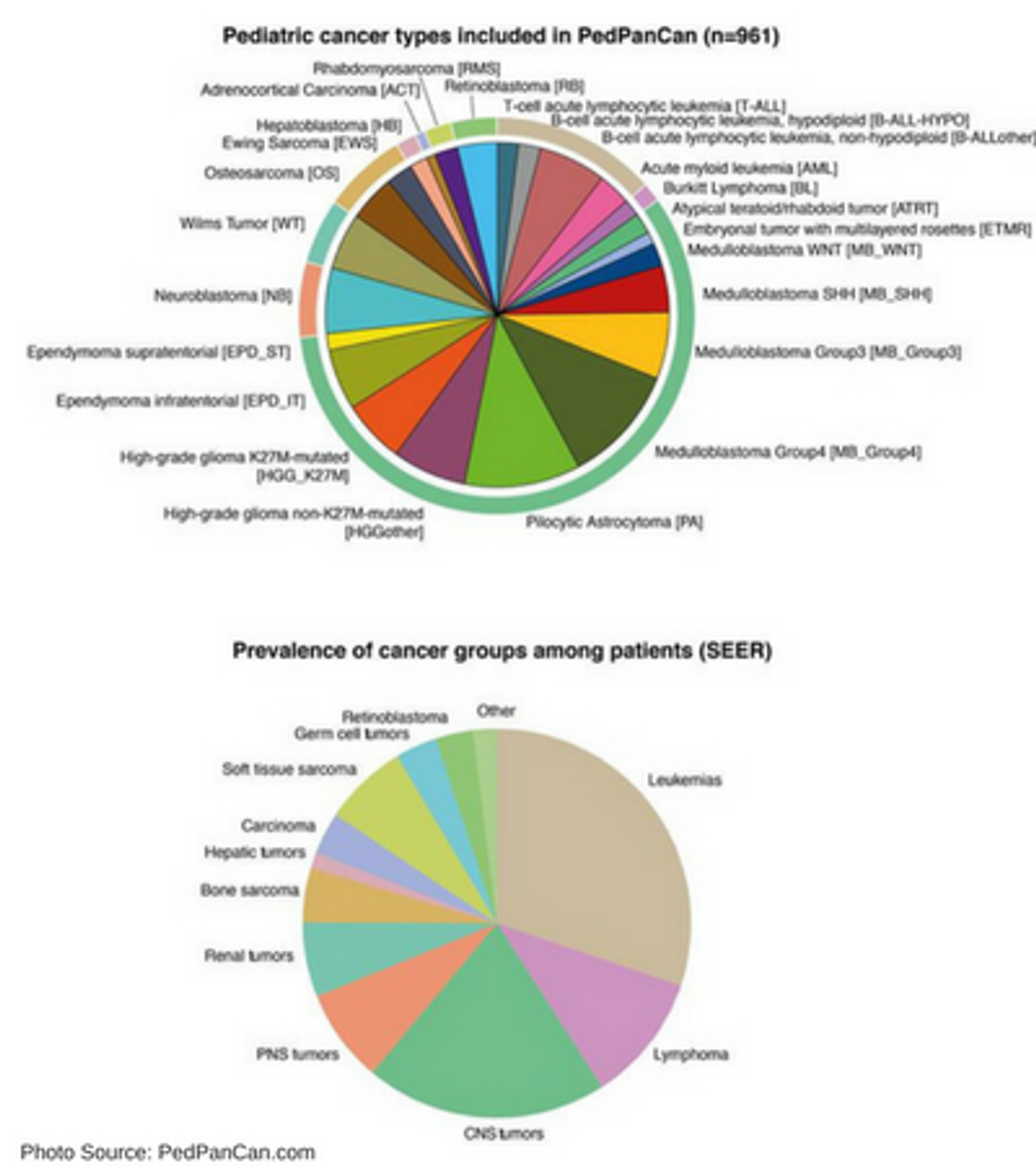 Two new studies published in Nature have evaluated pediatric cancers broadly across the genome in a way that has not been done before. The term “pan-cancer analysis” refers to the collection of molecular aberrations and identified commonalities or differences in the expression and regulation of biological processes in cancer cells from different lineages. The analysis includes somatic alterations, single nucleotide variants, insertions, deletions, structural variations, copy number alterations, gene fusions detected, and internal tandem repeats. Between the two studies published in late February 2018, the groups evaluated a total of 2,660 pediatric patient samples across a total of 30 types of molecularly or histologically different cancers.
Two new studies published in Nature have evaluated pediatric cancers broadly across the genome in a way that has not been done before. The term “pan-cancer analysis” refers to the collection of molecular aberrations and identified commonalities or differences in the expression and regulation of biological processes in cancer cells from different lineages. The analysis includes somatic alterations, single nucleotide variants, insertions, deletions, structural variations, copy number alterations, gene fusions detected, and internal tandem repeats. Between the two studies published in late February 2018, the groups evaluated a total of 2,660 pediatric patient samples across a total of 30 types of molecularly or histologically different cancers.
Ma, X. et al. found 142 pediatric cancer driver genes in their transcriptome analysis; only 45% of those found match drivers found in the TCGA adult pan-cancer analysis. They report that 62% of events are copy number alterations and structural variants. The median somatic mutation rate found for pediatric cancers (from 0.17 per megabase (Mb) in AML to 0.79 per Mb in osteosarcomas) differs greatly from the 1-10 mutations per Mb found in adult cancers.
Grobner, S. et al. focused more extensively on central nervous tumors but analyzed 24 types of cancer across body systems. They classified pediatric cancers into two categories based on 149 identified cancer driver genes. The small mutation classification focused on somatic mutations and data aligned with previous reports in the literature that generally somatic mutation burden increases as the patient ages with the exception of Burkitt’s lymphoma. The group reported that some cancers, like Acute Lymphoblastic Leukemia (ALL), Ewing’s sarcoma, and rhabdomyosarcoma had more random mutational loads. The second classification included cancers with structural or copy number variant changes including hyperdiploidy. The group found that many cancer types had more single nucleotide variants (SNVs). There were more point mutations that changed a cytosine (C) nucleotide to a thymine (T) nucleotide; however, findings also included transversions from cytosine (C) to adenosine (A). This group identified 30 known and 1 new mutational signature for each different tumor type.
Another interesting finding was that there is a 50% hereditary predisposition for adrenocortical carcinoma and 28% hereditary predisposition for hypodiploid B cell-ALL. New putative predisposition genes LZTR1, TSC2 and CHEK2 were also detected. These germline variant changes are related to DNA repair genes when mismatches occur during replication or double stranded breaks need repair. Grobner, S. et. al, wrote, “Carriers of TP53 germline mutations (Li-Fraumeni syndrome), most common in adrenocortical carcinomas, hypodiploid B-ALL, SHH medulloblastomas, and K27wt high-grade gliomas, are at a 50% risk for early onset cancer compared to 1% overall…”.
There is so much information and data available; next steps must include additional efforts to structure the information to be clinically relevant for patient diagnosis, prognosis, and future treatment research.
Sources: Nature, doi:10.1038/nature25480; Nature, doi:10.1038/nature25795, German Cancer Research Center DKFZ & Hopp Children’s Cancer Center, PedPanCan,