Parkinson’s disease is the second most common neurodegenerative disease, currently affecting ~ 1 million people in the United States. Just as with Alzheimer’s disease (the most common type of neurodegenerative disease), there are currently no treatments to slow Parkinson’s disease progression. Parkinson’s disease (PD) can be genetic, sporadic, or caused by exposure to certain pesticides. The disease pathology consists of degeneration of the dopamine-producing neurons in the substantia nigra, a region of the brain where voluntary movement is initiated. Exactly why these specific neurons are vulnerable is a big question in the PD research field.

Another major question in the field is how two of the main disease pathologies are connected. PD is characterized by Lewy bodies, which are aggregates of a protein called α-synuclein, and by mitochondrial dysfunction. A recent paper published in
Science Translational Medicine has answered this question and characterized a mechanism that links α-synuclein and mitochondrial dysfunction.
Mitochondria are double membrane-bound organelles that produce the energy that is used by the cell. As you’ve probably heard in high school biology, they are the “powerhouse” of the cell. They have their own DNA, but it only encodes 13 proteins, and up to 1,500 unique proteins are required for mitochondrial function. That means that efficient protein import is vital for mitochondrial function.
Researchers at the University of Pittsburgh School of Medicine have discovered that certain species of post-translationally modified α-synuclein bind to a mitochondrial import protein called TOM20 and that interaction leads to deficient respiration, increased production of reactive oxygen species (ROS), and a loss of mitochondrial membrane potential, all hallmarks of dysfunctional mitochondria.
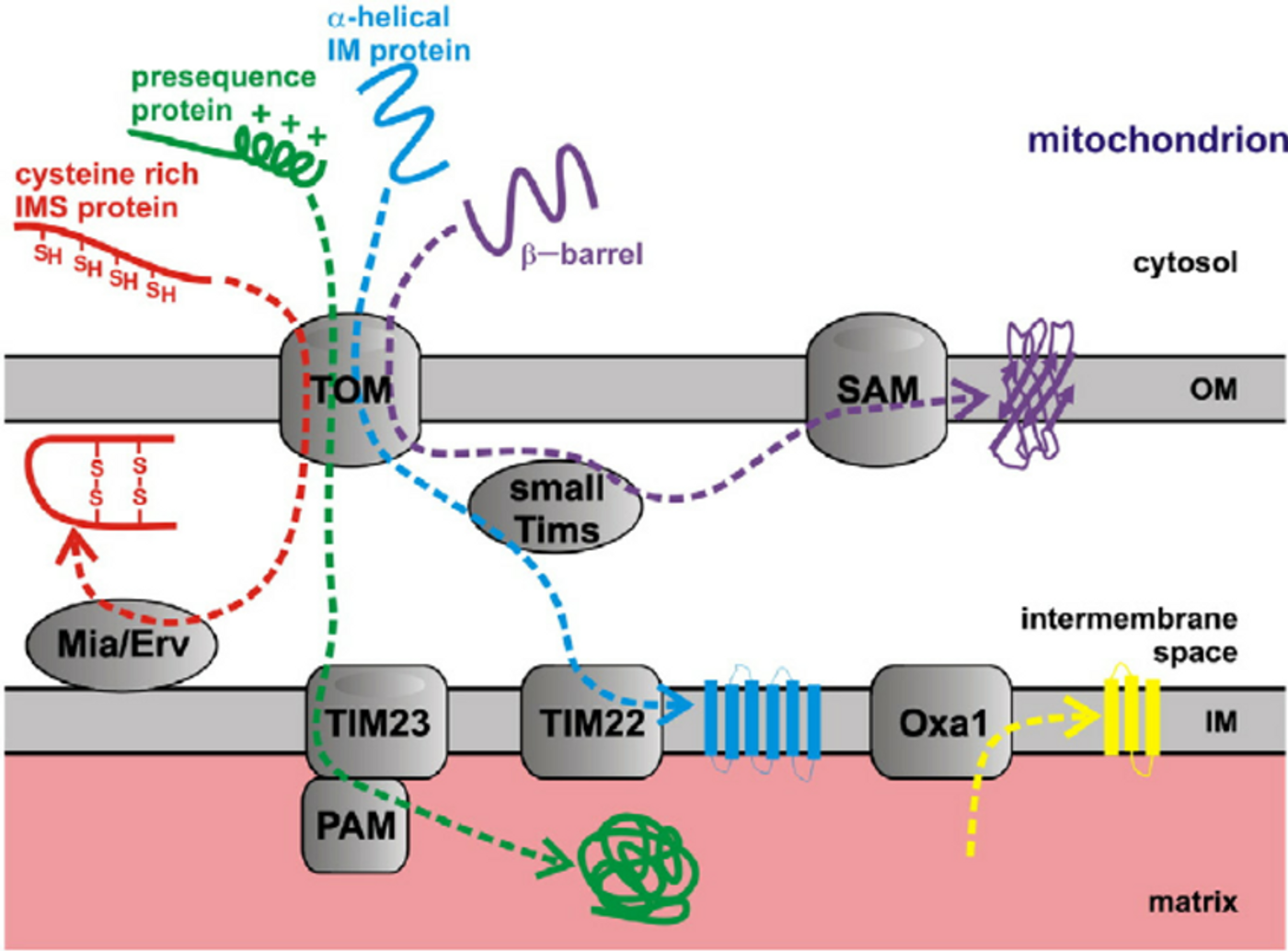
When a protein is targeted to the mitochondria for import, it contains a mitochondria targeting signal (MTS) that is cleaved once inside the mitochondria to produce the mature, functional protein. The MTS motif has a very specific secondary structure in that it has a hydrophobic side and a hydrophilic side. The hydrophobic side interacts with TOM20 and then the hydrophilic side associates with a co-receptor, TOM22, before being translocated to TIM proteins on the inner membrane. The TIM proteins then translocate the MTS-tagged protein into the mitochondrial matrix. Certain post-translationally modified α-synuclein peptides adopt a conformation similar to known MTS motifs, bind to TOM20, prevent its interaction with TOM22, and impair mitochondrial protein import.
This research is very exciting because it provides a link between α-synuclein and mitochondrial dysfunction and opens the door for new therapeutic strategies. It also helps to answer the question of why the dopaminergic neurons in the substantia nigra are so vulnerable. These neurons have been shown to express more α-synuclein than neurons in the surrounding midbrain and also have a higher state of oxidation to begin with. There is probably a destructive feed-forward cycle of excess post-translationally modified α-synuclein impairing mitochondrial protein import and causing more ROS to be produced which in turn causes cell-wide dysfunction. For therapeutics, the researchers overexpressed a naked MTS and saw that not only did it prevent α-synuclein binding to TOM20, but was also able to reverse the loss of the TOM20-TOM22 interaction caused by α-synuclein. The MTS is a small peptide and could potentially be developed as a disease-modifying PD therapeutic.
Sources:
EurekAlert,
Science Translation Medicine,
Vereen (image 1 source), and
ResearchGate (image 2 source)